Kriegler-Nayar-oireyhtymä

Crigler-Najjar oireyhtymä — geneettinen häiriö luokan fermentopathia tunnettu rikkoo yksi linkki prosessissa neutralointi ja erittymistä bilirubiinin — konjugaation. Tämän tilan oireet ovat maksan synnynnäisten keltaisuus ja vaikeat neurologiset häiriöt, jotka voivat johtaa kuolemaan infuusion aikana. Kriegler-Nayar-oireyhtymän diagnosointi tapahtuu biokemiallisten testien avulla ja konjugoimattoman bilirubiinin määrän määrittämiseksi veriplasmassa sekä molekyyligeneettisiin menetelmiin. Erityisiä taudin hoitoon ei ole olemassa (paitsi maksansiirtoa) hoito on lisätä tuhoaminen ja poistaminen bilirubiini erittyy (hemosorption, valohoitoa, plasmafereesiä ottaen barbituraatit).
- Kriegler-Nayar-oireyhtymän syyt
- Kriegler-Nayar-oireyhtymän luokitus ja oireet
- Kriegler-Nayarin oireyhtymän diagnosointi
- Kriegler-Nayar-oireyhtymän hoito
Kriegler-Nayar-oireyhtymä
Crigler-Najjar oireyhtymä — vakava geneettinen häiriö ominaista häiriö konjugoinnin bilirubiini glukuronihapon kanssa, joka on tärkeä vaihe sen hävittäminen ja poistaminen kehosta. Ensimmäistä kertaa tauti kuvattiin vuonna 1952 kaksi amerikkalaista lastenlääkärit — John Crigler Najjar ja Viktor. Lisätutkimus Crigler-Najjar syndrooman on osoittanut, että tämä ehto on geneettinen luonne ja peittyvästi periytymistapa lisäksi pystyimme tunnistamaan kaksi kliinistä muunnelmia tauti. Sairaus on suhteellisen harvinaisia, joten tarkkoja lukuja esiintyminen ei ole määritelty — enemmistön tutkijat uskovat, että se on tasolla 1: 1 000 000. sukupuolijakauma sairastavien potilaiden Crigler-Najjar syndrooman ei ole erikoisen tauti yhtä usein vaikuttaa sekä pojille sekä tytöt että tytöt. Tämän tilan hoidossa varhainen (ihanteellinen — prenataalinen) diagnoosi on äärimmäisen tärkeä, koska sairauden ennuste ja potilaan elämänlaatu ovat suuresti riippuvaisia hoidon alkuvaiheen ajallisuudesta.
Kriegler-Nayar-oireyhtymän syyt
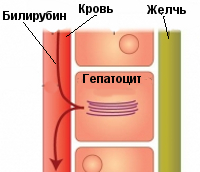
Crigler-Najjar oireyhtymä viittaa luokan fermentopathia (toinen luokitus — ryhmä konjugoimattoman hyperbilirubinemian), syy on siinä vajaatoiminnan sairauksien uridindifosfatglyukuronidazy 1, jonka tehtävänä on sitoa bilirubiini, jossa on kaksi molekyyliä glukuronihapon. Seurauksena tämä biokemiallinen prosessi bilirubiini voi liueta veteen, näkyy koostumuksen sapen ja, mikä tärkeintä, sen myrkyllisyys laskee merkittävästi. Kun Crigler-Najjar oireyhtymä, tämä prosessi dramaattisesti hidastunut tai sitä ei ole ollenkaan, mikä aiheuttaa viivettä poistaminen bilirubiini kehosta ja sen kerääntyminen. Bilirubiini on korostunut neurotoksisuuden suuremmilla pitoisuus veressä alkaa tallettaa aine kudoksissa ihon ja limakalvojen, mikä johtaa kehitystä keltaisuutta. Kun bilirubiini pitoisuus ylittää tietyn kynnyksen, yhdiste alkaa tunkeutua veri-aivoesteen aivojen, mikä johtaa vakavaan tautiin (erityisesti vaurioitunut pohjapinta ydin). Hoitamattomat potilaat Crigler-Najjar syndrooman kuolee lukuisten neurologisten sairauksien ja lisäämällä maksan koomaan.
Syynä vähäisen aktiivisuuden uridindifosfatglyukuronidazy mutaatiot UGT1A1-, joka sijaitsee kromosomissa 2, on vastuussa aminohapposekvenssi ja ottamalla entsyymi talteen. Lukuun ottamatta Crigler-Najjarin oireyhtymä Viat tämän geenin voi johtaa useita muita häiriöitä bilirubiini aineenvaihdunnan perinnöllinen luonne — Gilbert -oireyhtymä, ohimenevä vastasyntyneen bilirubinemia perheen tyyppi. Mekanismi perintö UGT1A1- geenin mutaatiot Crigler-Najjar oireyhtymä, peittyvästi periytyvä. Seuraavassa kuvataan useita tämän geenin mahdollisen vahingon muunnoksia, jotka johtavat taudin eri kulkuun.
Kriegler-Nayar-oireyhtymän luokitus ja oireet
Tällä hetkellä on kuvattu Kriegler-Nayar-oireyhtymän kahta pääasiallista kliinistä muotoa, jotka poikkeavat pääasiassa ilmentymien vakavuudesta ja taudin ennusteesta. Tämä johtuu geneettisen virheen tyypistä UGT1A1: ssä. Ensimmäinen sairauden tyyppi (SKN-1) aiheutuu missense-mutaatioista, jotka johtavat alempaan entsyymiin, jolla on intrasellulaarisen proteiinin käyttökelpoisuuden omaavien aminohappojen signaalisekvenssi. Siten tämän muodon avulla geenin puute vaikuttaa koodaaviin alueisiin (eksonit), mikä aiheuttaa patologian kehittymistä homotsygootteissa. Pian muodostumisensa jälkeen uridiinidifosfaattiglukuronidaasi 1 tuhoutuu ja bilirubiinin konjugoitumista ei tapahdu lainkaan.
Krigler-Nayyar-tyypin 1 oireyhtymälle on tunnusomaista vakava ja nopea virta — ensimmäiset keltaisen keltaisen hyperbilirubinemian merkit ovat vain muutama tunti syntymän jälkeen. Ajan myötä heihin liittyy neurologiset häiriöt — nystagmus, kouristuskohtaukset ja joskus opisthotonus. Keltaisuus pysyy koko lapsen elämässä, hänen henkinen kehitys on pienempi kuin hänen ikäisensä, taudin oireet kasvavat tasaisesti jopa intensiivisen hoidon aikana. Yleensä potilaat, joilla on Kriegler-Nayyar-tyypin I oireyhtymä, kuolevat ensimmäisen elämänvuoden aikana bilirubiinimyrkytyksen ja basaalisen subkorttisen ydinvaurion (ydin enkefalopatian) vuoksi.
Syy Crigler-Najjarin oireyhtymä tyypin 2 ovat myös missensemutaatioita UGT1A1- geeni, mutta ne voivat esiintyä koodaavan sekvenssin (eksonit) tai promoottori — osa on vastuussa geenin ilmentymistä. Useimmilla SCN-2-potilailla on defek- si-eksoni yhdellä kromosomilla, ja toisaalta promoottori, toisin sanoen tällaiset yksilöt ovat yhdiste heterotsygootteja. Tuloksena on vastoin viallisen entsyymin muotoa, tuotteiden uridindifosfatglyukuronidazy, joka ei tuhoudu, vaan on pienempi (20-25% normaalista) toiminnallinen aktiivisuus. Siksi tyypin 2 Krigler-Nayyar-oireyhtymälle on ominaista vähemmän vakava kliininen kuva ja suotuisampi ennuste.
Ensimmäisten kuukausien ja vuosien sairastavien potilaiden elämänlaatua Crigler-Najjarin oireyhtymä tyyppi, joka usein esiintyy vain vähäisiä keltaisuus, neurologiset poikkeavuuksia saattaa kehittyä ilman hoitoa nuoruusiässä. Useissa tapauksissa, erityisesti oikein määrätyillä terapeuttisilla toimenpiteillä, ei ole lainkaan ristiriitoja keskushermostosta. Ilmentymiä keltaisuus lievempiä potilailla, joilla Crigler-Najjarin oireyhtymä tyypin 2 voi jatkua läpi elämän ja pidetään usein mittarina komplikaatioita ja pahentaa potilaan tilaa. Ikää, joskus nystagmia, kohtauksia voidaan kirjata, mutta taudin oireiden kurssi ja vakavuus riippuvat täysin hoidon laadusta ja asiantuntijoiden suositusten täytäntöönpanosta.
Kriegler-Nayarin oireyhtymän diagnosointi
Kriegler-Nayyar-oireyhtymän diagnosointi perustuu lapsen yleiseen tutkimukseen, veren, sapen ja virtsan biokemiallisiin tutkimuksiin sekä molekyyligeneettisiin analyyseihin. Käytössä tutkimus paljasti keltaisuus, joka syntyi aamuyöllä (at SKN-1) tai kuukauden (SKN-2) elonmerkkejä neurologisia häiriöitä (opisthotonos, nystagmusta, säilyvyyskoe ohimenevä heijastuksia). Potilailla, joilla on toinen Kriegler-Nayyar-oireyhtymä, neurologiset häiriöt voidaan rekisteröidä aikuisikään, kun taas lapsilla on vain keltaisuutta. Myös ikä voi liittyä sellaisiin ilmenemismuotoihin kuin neuroensensorinen kuurous tai koreoatetoosi.
Kun veren biokemialliset tutkimus paljasti vakavan hyperbilirubinemian epäsuora (jopa 200-350 mikromol / l), ei (ja Crigler-Najjarin oireyhtymä tyyppi 1), tai jyrkkä lasku pitoisuuden suoran bilirubiinin. Tämän yhdisteen konjugoitunut fraktio puuttuu sapessa SCN-1: n kanssa ja sitä on läsnä pienissä määrissä SKN-2: n kanssa. Fenobarbitalovaya näyte Crigler-Najjarin oireyhtymä on positiivinen vain, jos uridindifosfatglyukuronidazy, eli kun SKN-2. Konjugoituneen bilirubiinin pitoisuuden tutkiminen virtsassa osoittaa sen kasvavan. Molekyyligeneettinen diagnoosi oireyhtymä Crigler-Najjar teki perinnöllisyyslääkärin — hän tekee suoralla sekvensoinnilla UGT1A1- geenin sekvenssin mutaatioiden havaitsemiseksi. Perinnöllisestä taudista, jonka perinnö on tästä taudista, vanhemmat voivat joutua patologian synnynnäiseen diagnoosiin. Differentiaalinen diagnoosi olisi tehtävä tavallisella vastasyntyneiden ohimenevän keltaisuuden ja Gilbertin oireyhtymän varalta.
Kriegler-Nayar-oireyhtymän hoito
Erityistä hoitoa tai etiotrop oireyhtymä Crigler-Najjar nykyään ei ole olemassa, kaikki hoitotoimenpiteet määrätty nopeuttaa hajoamista bilirubiinin ja sen erittymistä elimistöstä ja suojella keskushermostoon. Erityiset eroja hoidon 1. tai 2. sairauden laadusta ei ole läsnä (paitsi aktivointi mikrosomaalisiin hapettumisen barbituraatit, jota ei suoriteta 1st tyyppi), mutta SKN-1 hoito vain hieman puhkeamisen viivyttämiseksi kuoleman. Kaikkein radikaali menetelmän hoitaa Crigler-Najjarin oireyhtymä on tällä hetkellä allotransplantaation leikkaus maksan suhteellinen tai geneettisesti samankaltaisia luovuttajan — runko on muodostettu uridindifosfatglyukuronidazy.
Crigler-Najjarin oireyhtymä tyypin 2 käsitellään nimeäminen kohtalainen annoksia barbituraatit aktivoida hapettamalla bilirubiinin ja lisäämällä muodostumista haluttua entsyymiä. Lisäksi plasmapereesi, hemosorptiota ja verensiirtoa osoitetaan — kaikki nämä menetelmät on tarkoitettu eliminoimaan konjugoitunut bilirubiini elimistöstä. Hyviä tuloksia potilailla, joilla on Crigler-Najjarin oireyhtymä antaa valohoitoa — ihon altistumisesta johtaa osittainen tuhoutuminen bilirubiinin ja vapauttamaan kudosta reseptorien uusia osia toksiinin, mikä vähentää sen pitoisuus veressä. Oikea juominen ja lisääntynyt nesteenotto nopeuttavat toksiinin poistamista kehosta, joten vältä kuivumista. On välttämätöntä seurata jatkuvasti tämän aineen pitoisuutta veriplasmassa,
Kriegler-Nayarin oireyhtymän ennuste ja ehkäisy
Ennuste oireyhtymä Crigler-Najjarin tyypin 1 erittäin huono — koska täydellinen puuttuminen Entsyymiaktiivisuuden uridindifosfatglyukuronidazy 1 potilaista kuolee ensimmäisen elinvuoden komplikaatioista johtuva ydinaseiden tautiin. Aikana SKN-2 riippuu sellaisista tekijöistä kuin vakavuus ilmenemismuotoja, diagnoosi ja ajoissa hoidon aloittamista, suositusten noudattaminen asiantuntijoiden, läsnäolo tai puuttuminen on muita sairauksia. Useimmissa tapauksissa suhteellisen suotuisa ennuste — potilaalla on Crigler-Najjarin oireyhtymä tyypin 2 voi säilyä jopa vanhuuden, luonteenomaisen ilmenemismuotoja tauti voi vain häiritä keltaisuutta. Ehkäisy tämä ehto on mahdollista vain puitteissa genetiikan kuulemisen vanhemmille, jotka ovat suvussa tämä sairaus, sekä avulla sikiödiagnostiikkaa.