Gaucherin tauti
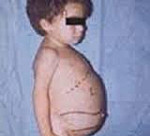
Gaucherin tauti – geneettinen sairaus, jolle on tunnusomaista lipidimetabolian rikkominen, lysosomaalisen entsyymin puutos, glykolipidien kerääntyminen solurakenteisiin. Oireet määräytyvät patologian tyypin mukaan. Yleisiä oireita ovat maksan laajentuminen, perna, veren hyytymistä vähentynyt. Kun tyyppi I paljastui luustoelinten rikkomukset: osteoporoosi, usein murtumia, luu-infektioita. Tyypillisessä tyypissä II ja tyypissä III hallitsevat neurologiset oireet: kouristukset, halvaus, karsastus, henkinen hidastuminen. Diagnoosi perustuu puutosentsyymin biokemialliseen analyysiin. Hoito sisältää entsyymien korvaamisen, substraatti vähentää ja oireenmukaista hoitoa.
- Gaucherin taudin syyt
- synnyssä
- Gaucherin taudin oireet
- komplikaatioita
- diagnostiikka
- Gaucherin taudin hoito
- Ennuste ja ennaltaehkäisy
Gaucherin tauti
Sairaus sai nimensä ranskalaisen lääkärin Philippe Gaucher nimeksi. Vuonna 1882 hän kuvaili potilaan pernan rakenteen oireita ja patogeenisia piirteitä, joka kuoli sepsiksestä. Useita vuosikymmeniä myöhemmin, samanlaisessa kliinisessä tapauksessa, Gaucher määritteli glukoserebrosidin kerääntymisen pernassa ja glukoserebrosidaasin entsyymi. Gaucherin tauti (sfingolipidoz, glukosyylikeraami lipidoosi) kuuluu lysosomaalisten kerääntymisvaurioiden ryhmään – perinnölliset sairaudet, jonka mukaan lysosomien solujen organelujen tehtävät muuttuvat. Taudin taajuus on 1:40 tuhatta. enintään 1:70 tuhatta. Yleisyys on suurin yhteisössä, jossa lähisukulaisten väliset avioliitot sallitaan, esimerkiksi, Ashkenazi-juutalaisia. Mutaatiogeenin kuljettaminen määritetään noin yhdellä henkilöllä 400: sta.
Gaucherin taudin syyt
Glykosyyliseramidi sfingolipidoosi on yleisimpiä perinnöllisiä entsyymipatioita. Kehityksen syytä pidetään GBA-geenin virheenä, joka koodaa lysosomientsyymi beeta-glukosidaasia (glukoserebrosidaasia), joka on vastuussa lipidien hajoamisesta. Taudin perintö tapahtuu autosomaalisella resessiivisellä tavalla, muutetun geenin läsnäolo on välttämätöntä fermentopatian muodostumiselle: yksi – äidiltä, muut – isältä. Avioparissa, missä molemmat vanhemmat ovat – mutaatio kantajat, sairastuneen lapsen todennäköisyys on 25%. Riskin yhden viallisen geenin lähettämisestä, eli kuljetuksen riski kehittämättä sairautta tällaisissa perheissä on 50%. Geenitekniikan kahden mutantti-alleelin läsnä ollessa glukoserebrosidaasifunktio pienenee 15-30% normaalista tasosta.
synnyssä
Taudin patogeeninen perusta on beta-glukosidaasin katalyyttisen aktiivisuuden väheneminen. Tämän seurauksena glykosfingolipidien pilkkoutumisprosessi häiriintyy (monimutkaisten lipidien ja hiilihydraattien yhdisteet) glukoosiksi ja ceramidiksi. Soluissa tapahtuu makromolekyylien epänormaalia progressiivista kerääntymistä, joiden ominaispiirteet ovat päivityksen nopeutettu nopeus – makrofagissa. Ei-hydrolysoidut lipidit konsentroidaan lysosomeihin, muodostuu erityisiä kerääntymiskennoja – Gaucher-solut. Ensisijainen aineenvaihduntahäiriö herättää toissijaisia häiriöitä biokemiallisissa prosesseissa ja solukkotoiminnoissa. Rasvan aineenvaihdunnan patologian takia makrofagin aktivaatio -oireyhtymä kehittyy. Stimuloidaan monosytoopiisilla, lisääntynyt makrofagien sisältö maksassa, perna, luuytimen. Tämä aiheuttaa splenomegalia, hepatomegalia, luuytimen infiltraatio. Makrofagien säätelytoiminnan häiriö on sytopenian aiheuttaja, luiden ja nivelten vaurioita.
Gaucherin taudin oireet
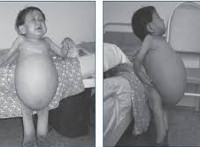
Debyyttialalla ja kliinisillä ominaisuuksilla on kolmenlaisia sairauksia. Ensimmäinen tyyppi on yleisimpiä, on krooninen kurssi. Oireita esiintyy usein 30-40 vuotta, harvemmin, tauti ilmenee lapsena. Maksan ja pernan koon kasvu alkaa välittömästi syntymän jälkeen, mutta kliinisesti ilmenee myöhemmin. Ensimmäiset patologian merkit ovat anemia, lisääntynyt verenvuoto. Hematopoieettisen järjestelmän inhibitioon liittyy hemoglobiinin ja verihiutaleiden määrän väheneminen. Tuki- ja liikuntaelimistön muutoksia edustaa kipu luissa ja nivelissä, usein murtumia, muodonmuutokset (yleensä, reisiluun muutokset). Kasvojen ja jalkojen hyperpigmentaatio on havaittavissa aikuisilla: iho pimenee, hankkii sävy kellertävästä kelta-ruskeaksi. Litteitä punaisia pilkkuja voi esiintyä tyypillisellä lokalisoinnilla silmien ympärillä. Potilaskorkeus alle keskiarvon.
Toinen taudin tyyppi (akuutti lapsi tai akuutti neuropaattinen) hyvin harvinainen, kehittyy syntymästä puolitoista vuoteen, useimmiten oireet debytoivat kolmen ensimmäisen kuukauden aikana. Sillä on ominaista nopea virta, heikko hoitovaste. Neurologiset häiriöt tulevat esiin, joka aiheutuu Gaucherin solujen kertymisestä keskushermostoon. Lapset huutavat heikosti, hidas imee. Rikkoutunut hemmotteleva refleksi, usein epänormaali hengitysjakso. Henkisestä ja fyysisestä kehityksestä on huomattava viivästys. Taudin alkuvaiheessa lihasäänet vähenevät, 9-12 kuukautta debyyttien hypertonin esiintymisen jälkeen, varsinkin niskan ja raajojen lihaksissa. Spasmit kehittyvät, karsastus, spastinen halvaus. Maksa ja perna suurentuvat. Lapset kärsivät usein vakavasta keuhkokuumeesta.
Kolmas tyyppi – nuorena tai subakuutteina neuropaattisina. Ensimmäiset merkit – laajentunut perna ja maksa – syntyy 2-3 vuotta. Täydelliset oireet puhkeavat 6-15-vuotiaana. Keskushermosto-oireiden kliiniset oireet ovat lihasten hypertonia, spastinen halvaus, karsastus, tahattomia kouristuksia, kouristukset, hengitysvaikeudet hengitysvaikeuksilla, nielemisvaikeuksia. On psyykkisiä häiriöitä: henkisten toimintojen väheneminen, puheen ja kirjoittamisen puute, emotionaalinen epätasapaino, psykoosit. Lapset jäävät jälkeen seksuaalisessa kehityksessä. Taudin kulku on tasaisesti etenevä.
komplikaatioita
Vakavimmat komplikaatiot havaitaan taudin toisessa ja kolmannessa lajissa. Selkäydinnesteen ja aivojen vaurioituminen johtaa hengityselinten häiriöön, äkilliset äkilliset hengitysilmat, riski, että kurkunpään kouristus ja kuoleman tukehtuminen lisääntyvät. Pienentynyt verihiutaleiden määrä voi aiheuttaa suuren sisäisen verenvuodon. Ensimmäisessä tyypin patologiassa kärsivillä potilailla yleinen komplikaatio on luun tuhoaminen, niiden lisääntynyt epävakaus ja tartuntataudit. Rajoitettu liikkuvuus, potilaat eivät voi kävellä itsestään, tarvitsevat hoitoa.
diagnostiikka
Historian ottamista ja fyysistä tutkimusta suorittaa endokrinologi ja neurologi, Lisäksi nimitettiin neuvontaa genetiikka, hematologist, silmälääkäri, lastenlääkäri, psykiatri. Anamnistiset tiedot sisältävät Gaucherin taudin esiintymisen sukulaisilta. Tutkimus paljastaa tyypillisiä oireita: lyhyt korkeus, luupatologiat, neurologiset oireet (karsastus, ataksia, halvaus), verenvuotoinen oireyhtymä, ihon hyperpigmentaatio. Joskus sairauden epäilty tapahtuu suurentuneen pernan satunnaisen havaitsemisen jälkeen ultraäänikuvien yhteydessä, hematopoieettisen järjestelmän sortaminen täydellisen verenlaskun mukaan. Vahvistetaan diagnoosi, muiden metabolisten perinnöllisten patologioiden poissulkeminen, osteomyeliitti, luun tuberkuloosi, virusperäisen hepatiitin ja onkologisen verilasion erityisdiagnostiikka suoritetaan:
- kliininen, biokemiallinen veritutkimus. Trombosytopenia havaitaan suurimmalla osalla potilaista, leukopenia, anemia, joka lapsilla on yleensä raudanpuute. Biokemiallisen analyysin tulokset paljastivat alentuneen glukooserebrosidaasiaktiivisuuden.
- Entsyymisolujen analyysi. Gaucherin tauti kuivissa verinäytteissä ja ihon fibroblasteissa paljasti riittämätöntä glukosidaasin aktiivisuutta. Entsyymin puutosasteella ei ole suoraa korrelaatiota oireiden vakavuuden kanssa. Lisäbiokemiallinen merkkiaine – kitotriosidaasia. Tämä entsyymi syntetisoidaan aktivoitujen makrofagien avulla, tunnettu siitä, että sen aktiivisuus kasvaa 6-10 kertaa.
- Morfologinen tutkimus luuytimestä. Tautikohtaisten rakenteiden esiintyminen vahvistetaan – Gaucher-solut. Tuloksena eliminoituu hemoblastoosi ja lymfoproliferatiivinen sairaus.
- Luukudoksen rakenteen tutkimus. Osteo-nivelrakenteen vaurioiden vakavuuden arvioimiseksi suoritetaan densitometria, radiografia ja/tai MRI-luustoa. Mahdollinen diffuusi osteoporoosi, Erlenmeyer-pulloja voidaan visualisoida, osteolyysin foci, osteoskleroosi ja osteonekroosi. Osteopenia havaitaan taudin varhaisvaiheissa, luuytimen infiltraatio.
- Pernan kuvatutkimus, maksa. Ultrasound ja MRI sisäelimiä. Tulosten perusteella todetaan, että polttovälit ovat läsnä tai puuttuvat, suurennetun elimen tilavuus mitataan. Perusindikaattorit mahdollistavat myöhemmin hoidon tehokkuuden seurannan.
- Molekyyliset geneettiset tutkimukset. DNA-diagnostiikka on valinnainen menetelmä. GBA-geenin mutaation vahvistaminen on välttämätöntä, kun biokemialliset tutkimukset ovat epäselviä, sekä äitiys- ja implantaatiotutkimuksissa.
Gaucherin taudin hoito
Erikoistunut hoito potilaiden, joilla on ensimmäinen ja kolmas sairaus, pyrkii poistamaan oireet ja kompensoimaan primaarisen geenivirheen – lisätä puuttuvan entsyymin määrää, lisääntynyt glykosfingolipidien katabolia. Tyypin 2 patologian terapeuttiset toimenpiteet eivät ole riittävän tehokkaita, lääkärien ponnistelut vähennetään kliinisten ilmentymien helpottamiseksi – kipu, kouristukset, hengitysvaikeudet. Yleinen järjestelmä sisältää seuraavat alueet:
- Entsyymien korvaushoito. Tärkein hoito on elinikäinen entsyymien korvaushoito (LFT) käyttämällä rekombinanttia glukoserebrosidaasia. Tehokkuus on riittävän korkea – oireet pysähtyvät kokonaan, potilaiden elämänlaatu kasvaa. ERT on suositeltavaa kolmannen ja ensimmäisen tyypin taudissa. Lääkkeitä annetaan laskimoon. Usein infuusio aiheuttaa joskus tulehdussairauksia suonissa (laskimotulehdus).
- Substraatti vähentää hoitoa. Tämä suunta on uusi Gaucherin taudin hoidossa, suhteellisen laajalle Yhdysvalloissa ja Euroopan maissa. Tavoitteena on vähentää glykosfingolipidisubstraatin tuotannon nopeutta ja nopeuttaa kerääntyvien makromolekyylien kataboliaa. Lääkkeet ovat spesifisiä glukosyylikeramidisyntaasin inhibiittoreita. Menetelmä on tarkoitettu tyypin 1 sairaudelle, jossa on lieviä ja kohtalaisia oireita.
- Oireinen hoito. Osteoporoosin tapauksessa on määrätty monimutkainen hoito, mukaan lukien kalsiumlisät, D-vitamiini ja dieetio, runsaasti kalsiumia. Nämä toimenpiteet auttavat hidasta luukatoa, lisää luun lujuutta, estää murtumia. Luuston komplikaatioita varten käytetään analgeetteja (Tulehduskipulääkkeet), antibakteerinen hoito. Hermoston sairauksien oireet pysäyttävät epilepsialääkkeet, nootropami, lihasrelaksantit.
Ennuste ja ennaltaehkäisy
Suotuisat tulokset ovat todennäköisesti tyypin 1 sairauspotilailla – Monimutkainen terapeuttinen lähestymistapa mahdollistaa glukoserebrosidaasin funktionaalisuuden normalisoinnin, estää komplikaatioiden kehittymistä, välttämään vammaisuutta. Tyypin 3 ennuste riippuu taudin luonteesta, yksilön vastaus terapeuttisiin toimenpiteisiin. Tyyppi 2 on äärimmäisen vaikea ja päättyy potilaan kuolemaan. Ehkäisy tapahtuu raskauden suunnittelun ja sen alkuvaiheessa. Perheille suositellaan geneettistä neuvontaa, joilla on läheiset sukulaiset tämän patologian kanssa. Suurella riskillä, että mutaatio siirretään syntymättömälle lapselle, ensimmäisellä raskauskolmanneksella tutkitaan entsyymin määrää amnioottifluidissa, abortin kysymystä käsitellään.