glykogeenin
glykogeenin – perinnöllisiä sairauksia, jotka perustuvat entsyymien tuotantoon liittyvään geneettiseen puutteeseen, mukana hiilihydraattien aineenvaihdunnassa. Luonteenomainen yhteinen piirre – liiallinen glykogeenin kertyminen myosyytteihin, hepatosyyttejä ja muita kehon soluja. Hypoglykemian glikogenoosi-oireet, hepatomegalia, lihasheikkoutta, maksan, sydän, hengitysvaikeudet ja munuaisten vajaatoiminta. Diagnoosi sisältää veren biokemiallisen analyysin, morfologinen tutkimus liha- ja maksabiopsian materiaalista, entsyymiaktiivisuuden määritys, molekyyligeneettiset testit. Hoito perustuu kliiniseen ravitsemukseen, lääketieteellisten oireiden korjaaminen, joissakin tapauksissa tarvitaan leikkausta.
- Glykogenoosin syyt
- synnyssä
- luokitus
- Glykogenoosin oireet
- komplikaatioita
- diagnostiikka
- Glykogenoosi hoito
- Ennuste ja ennaltaehkäisy
glykogeenin
Glykogenoosi on tutkittu vuodesta 1910 lähtien. Vuosina 1928-29 kuvattiin tyypin I glyko- genaasin oireita – «glykogeenin varastointitauteja». Vasta vuonna 1952 oli mahdollista tunnistaa entsyymivirhe ja muodostaa yhteys oireiden kehittymiseen. Patogeneettisiä mekanismeja ja hoitomenetelmiä ei vielä täysin ymmärretä. Tähän mennessä on tunnistettu 12 tyyppistä glykogeenimuotoa, täysin tutkittu 9. Alhainen esiintyvyys, keskimäärin 1 tapaus on 40-68 tuhatta ihmistä. Epidemiologiset indikaattorit ovat samat molemmilla sukupuolilla, mutta X-resessiivisellä perinnöllä miehet sairastuvat useammin. Oireet esiintyvät vastasyntyneiden aikana tai varhaislapsuudessa, nykyistä useammin jatkuvasti etenevää.
Glykogenoosin syyt
Ainoa tekijä, jotka aiheuttavat glykogeenitautien kehittymistä, on geneettinen vika, mikä johtaa tietyn entsyymin riittämättömyyteen, vaihtaa glukoosia. Kaikki glyko- genoosit, lukuun ottamatta tyyppiä IX, periytyvät autosomaalisen resessiivisen periaatteen mukaisesti. Se tarkoittaa, että mutationalgeeni sijaitsee kromosomissa, ei liitetty lattiaan, taudin ilmeneminen on mahdollista vain, jos kukaan vanhemmista muuntuu perimättä – jossa on kaksi recessive alterated geeniä allelissa. Jos parin yksi geeni on viallinen, sitten toinen – hallitseva, normaali – antaa keholle tarpeeksi entsyymiä. Henkilö muuttuu glykogeenisyn kantajaksi, mutta ei sairas. Pareittain, missä molemmat ovat kumppaneita – harjoittajat, sairastuneen lapsen todennäköisyys on 25%. Tyypin IX glyko- genoosissa patologinen geeni sijaitsee sukupuolen X-kromosomissa. Hemizygous miehet ovat pari XY, aina sairas glykogeenisillä, siirtää vika kaikille tyttärillensä. Muuntion siirto todennäköisyydestä naaraspuolisesta kantajalta molempien sukupuolten lapsille on 50%.
synnyssä
Kaikkien glykogenoosien patogeeninen perusta – glykogeenin synteesin ja hajoamisen prosessin mahdottomuus, sen kertyminen kudoksiin. Glykogeeni on ainoa ruumiinpysyvä polysakkaridi, eräänlainen energia «varikko» – aterian jälkeen ylimääräinen glukoosi muunnetaan maksan ja lihasten glykogeeniksi, sitten vähitellen jakautuu takaisin glukoosiin. Tämän mekanismin ansiosta pysyvät verensokeriarvot säilyvät, solujen ja kudosten energiaa toimitetaan jatkuvasti. Kun aglykogenoosi (0-tyyppiä) – ei ole glykogeenisyntaasientsyymiä, glykogeenin tuotantopäällikkö. Potilaat kärsivät vakavasta hypoglykemiasta.
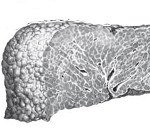
Kun tyypin 1-11 glikogeeni-tautia esiintyy minkä tahansa entsyymin geneettisesti määritelty puute, katalysoimalla glukoosi-glykogeeniglukoosin ketju. Tyypille 1 on tunnusomaista glukoosi-6-fosfataasin ja glukoosi-6-fosfaatti-transloakkeen, Tyyppi 2 – alfa 1,4-glukosidaasi, Tyyppi 3 – AMILO-1,6-glukosidaasi, Tyyppi 4 – D-1,4-glukaani-α-glukosyylitransferaasin, Tyyppi 5 – myosyyttiglykogeenifosforylaasi, Tyyppi 6 – hepatosyyttärkkelysfosforylaasi, Tyyppi 7 – fosfoglukomutaasi, Tyyppi 8 – fosfofrukto-mutaasin, Tyyppi 9 – hepatosyytti fosforylaasikinaasi. Aktiivisuuden vähenemisen tai entsyymien täydellisen puuttumisen vuoksi glykogeeni kerääntyy lihaksiin, maksa, harvoin – muissa kudoksissa. Elinten rakenne ja toimivuus muuttuvat, kehittyy erilaisia organismin vikoja.
luokitus
Ottaen huomioon entsymaattisen puutteen ja kliinisten ilmentymien piirteet, erotetaan 12 glyko- genaasivarianttia, 0: sta xi: een. lisäksi, kuvataan yhdistettyjen tyyppien tapauksia, kun määritetään kaksi entsyymien puutetta, samoin kuin tunnistamattomia tyyppejä, jossa ei voida eristää entsyymivirhettä. Johtavan patogeneettisen mekanismin mukaan glykogeenitaudit jaetaan kolmeen suureen ryhmään:
- maksan. Sisällytä kaikentyyppiset glykogeenit, paitsi II, V ja VII. Glykogeeni on talletettu pääasiassa hepatosyytteihin. Hepatomegaly on ominaista, hypoglykemia 2 tuntia hiilihydraattien nauttimisen jälkeen. Kun tyypin I tauti vaikuttaa myös munuaisiin, myopatiat kehittyvät tyypillä III ja IV.
- lihas. Tämä ryhmä sisältää tyypin VII ja V sairauksia. Muutettu entsyymiaktiivisuus lihaskudoksessa, rikki lihasvoimaa. Tyypillisiä oireita – lihaskipu, kouristukset.
- hybridi. Tyypin II glykogenoosi on erilainen siinä, että kaikki glykogeenipitoiset kudokset osallistuvat patologiseen prosessiin. Glykogeeni kerääntyy lysosomeihin ja solusytoplasmaan. Monet elimet kärsivät, lisääntynyt sydämen tai hengitysvajeen aiheuttaman kuoleman riski.
Glykogenoosin oireet
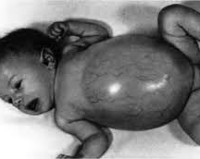
Aglycogenosis kehittyy vastasyntyneiden tai varhaislapsuuden aikana. Pieni glykogeenipitoisuus maksassa ilmaantuu voimakkaalla paaston hypoglykemialla. On hidastumassa, syvän unen, tajunnan menetys, vaalea iho, pahoinvointi, oksentelu, kramppeja yöllä ja aamulla. Ulkopuolella potilaat ovat tukkeutuneita, on pienempi luun tiheys, lisääntynyt murtumisriski. Girke-taudin kanssa (Tyyppi I) oireet debytoivat ensimmäisen neljän kuukauden aikana. Havaittu huono ruokahalu, oksentelu, painon puute, suurentunut maksa, kehonrakenteen epäsuhtaisuus – pyöreät kasvot, iso vatsa, ohuet raajat.
Pompe-taudin kliiniset oireet (Tyyppi II) määritetään muutaman viikon kuluttua syntymästä. Hitaat lapset, istuma-, heikentyneen imusrefleksin kanssa, vähentynyt ruokahalu. Hepatomegaly muuttaa kehon mittasuhteita – vatsa kasvaa, kädet ja jalat ovat ohut. Sydän on iski, keuhkot, hermostoa. Sydämen ja keuhkojen vajaatoiminnan suuri riski. Potilailla, joilla on Forbes-tauti (Tyyppi III) oireet lievästä tai kohtalaiseen vakavuuteen. Imeytymisen jälkeinen hypoglykemia tulee esiin, hepatomegalia, ihonalaisen rasvan kertyminen kehon alueelle. Andersenin taudin johtavat oireet (Tyyppi IV)– lihasheikkoutta, huono harjoittelutoleranssi, kouristukset.
Thomsonin tautia edustaa hepatomegalia, silmävärve, ataksia, progressiiviset neurologiset häiriöt lihasten kohonnut verenpaine, decerebration. MacDardin taudin tyypilliset ilmentymät (V-tyyppi) – kivut, keuhkopussin supistukset, liiallinen väsymys ja lihasten heikkous pienen kuorman jälkeen. Joskus tonic kouristukset yleistyvät, johon liittyy yleinen jäykkyys. Gersin taudin ilmenemismuodot (VI-tyyppiä) vähemmän voimakas, potilaat pystyvät sietämään kevyttä ja kohtalaista liikuntaa, ilman epämukavuutta. Lisäksi havaitaan maksavaurion oireita – ruokahaluttomuus, oksentelu, pahoinvointi, kipu oikealla puolella.
Taudin tauti Tarui (Tyyppi VII) sisältää liikunta-intoleranssia, johon liittyy pahoinvointia ja oksentelua, kivuliaita lihaskramppeja. Glukoosin saanti ei lisää kykyä suorittaa fyysisiä toimenpiteitä. Ruokien jälkeen oireet pahenevat. Hagan taudin kaikkein hyvänlaatuinen kulku (Tyyppi IX). Sairaat kasvavat maksan, viivästyttää moottorin kehitystä ja kasvua, hypotensio muodostuu. Oireita vähennetään iän mukaan. Tyypin X glykogenoosi on erittäin harvinaista, tunnusomaista hepatomegalia, pitkäaikainen harjoittelunsietokyky. Tyypin XI glyko- genoosiin liittyy merkittävä maksan lisääntyminen, kasvun hidastuminen ja fyysinen kehitys, riisitauti. Nuorilla maksan tilavuus on usein vähentynyt, kasvun kiihtyvyys.
komplikaatioita
Glykogeenin lajikkeilla, johon liittyy hypoglykemia, hypoglykeeminen kooma voi kehittyä. Yleensä, veren glukoosin voimakas väheneminen tapahtuu aterioiden ohittamisessa, varsinkin kun yön unta (ohita aamiainen). Potilaat kokevat huimausta ja kouristuksia, menettää tajuntansa. Vaikeat lihasglykogeenihoidon muodot, joilla on pitkittynyt hoito ja hoidon puuttuminen, johtavat luuston lihasten dystrofiaan, sydämen vajaatoiminta. Tiettyjen maksasiglykosgeenien komplikaatio on kirroosi.
diagnostiikka
Jos epäillään glykogenoosia, lapsen on suositeltavaa kuulla genetiikkaa, lastenlääkäri, gastroenterologist, hepatologist. Ensinnäkin erikoislääkäri kerää anamneesin, suorittaa kliinisen tutkimuksen ja tutkimuksen. Koska tauti on siirretty autosomaalisella resessiivisellä tavalla, perhetapauksia harvoin havaitaan. Heikkouden yhteiset valitukset, lapsen apatia, vaalea ja keltainen iho, kieltäytyminen syömisestä tai ruokahalun lisääntyminen, vaikeuksia aamulla aamulla, vapina, kouristukset. Tutkittaessa lääkäri huomauttaa maksan koon kasvaessa, vatsan pullistuminen, kasvun hidastuminen, lihaksen hypotrofia, ihonalaisen rasvan kertyminen, keltakasvamaan. Laboratorio- ja instrumentaalimenetelmät mahdollistavat glykogeenin diagnoosin vahvistamisen, poista synnynnäinen kuppa, toksoplasmoosi, cytomegaly, maksa-patologia, Gaucherin tauti, myotonia, progressiivinen lihasdystrofia, amyotrofiakompleksia. Pakollisia tutkimusmenetelmiä ovat mm:
- Biokemiallinen veritesti. Analyysin tulokset osoittivat hypoglykemiaa paastoglukoositasolla 0,6-3 mmol/l, maitohappoasidoosi, jonka maitohapon pitoisuus on 3-10 mmol/l (paitsi glykogeenisyystyyppi 4). Lisäksi triglyseridien lisääntyminen, kokonaiskolesteroli, LDL, VLDL, virtsahappo, maksan entsyymit.
- Maksabiopsi, lihakset. Maksakudoksen tutkimuksessa yleisimmät ominaisuudet ovat lisääntynyt glykogeenin määrä ja sen glybinen jakautuminen hepatosyyttien sytoplasmassa, joskus – vakuolaatioytimissä. Määritetty voimakas proteiini ja/tai suuren ja pienen rasvaisen hepatosyyttien degeneraation, niiden nekroosi, fibroosin limakalvot solukuoltoon kohdissa. Kirroosin merkit ovat mahdollisia. Lihaksen biopsiaa tutkitaan lihaksen tyyppisissä sairauksissa, jossa rakenteellisen normaalin glykogeenin subsarkolemal kertymät ovat näkyvissä.
- Entsyymi-analyysi. Entsyymiaktiivisuutta tutkitaan ihon fibroblastiviljelmässä, lihasten ja maksakudoksen biopsia, valkosolut. Kun glykogeenisaatio on krooninen hitaasti etenevä kurssi, funktionaalinen entsyymin väheneminen on lievä tai kohtalainen. Vaikeissa tapauksissa entsyymi puuttuu tai sen aktiivisuus on vähäinen.
- Vatsan ultraääni. Maksa on merkittävästi lisääntynyt, varsinkin sen vasen lohko. Hyperokogeenisyys ja parenkyymin rakenteellinen diffuusi heterogeenisyys ovat ominaisia (useita pieniä hyperecoomaisia kaikuja, jaetaan tasaisesti). Distaalisessa parenkyymissä ultraäänen kulku heikkenee. Rakenteellisesti erilaisten maksan adenoomien havaitseminen on mahdollista, lisääntynyt munuaisten koko, perna ja haima.
Diagnostiikkatutkimusten monimutkaisuus valitaan erikseen potilaan iän ja glikogeeni-taudin tyypin mukaan. Molekyyligeneettistä diagnoosia voidaan vaatia (geenin sekvensointi mutaatioiden tunnistamiseksi), rheotachygraphy, ekokardiografia, OAK, coagulogram.
Glykogenoosi hoito
Erityisiä hoitomenetelmiä ei ole kehitetty. Patogeneettinen hoito toteutetaan konservatiivisesti, pyritään poistamaan hypoglykemia, metabolinen asidoosi, ketoosi, hyperlipidemia, hepatiarinen kompleksin ja maha-suolikanavan dysfunktion korjaus. Kehittämällä komplikaatioita (vakavia sisäelinten vahingoksia) suoritetaan kirurgiset toimet. Potilaiden hoito sisältää seuraavat ohjeet:
- Ruokavaliohoito. Yksilöllinen ravitsemussuunnitelma laaditaan mineraalisten sairauksien minimoimiseksi. Potilaita kehotetaan vähentämään rasvan määrää, sakkaroosi, fruktoosia ja galaktoosia vähentämään hyperlipidemiaa ja asidoosia. Ensimmäisen tyypin glykogenogeesi on määrätty ruokavalio, jolla on lisääntynyt hiilihydraattien saanti. Erityisesti, osoittaa maissitärkkelyksen käyttö hidas sulavuutta, estävä hypoglykemia. Tyyppien 3 kanssa, 4 ja 9 tuo esiin ruokavaliota, jolla on enemmistö eläinproteiinista ja jakeittain ravinnosta.
- Oireiden huumeiden korjaus. Monimutkaisen käsittelyn puitteissa kokkarboksylaasia käytetään asetyylikoentsyymi A: n tuotannon lisäämiseen, kortikosteroidit ja glukagoni stimuloimaan glukoneogeneesiä. Carnitiinipuutosta korvaa vasen karnitiini. Toissijaisissa tubulopatioissa, maksan ja sappihäiriöiden hoitoon käytetään koleroeteettisia lääkkeitä, gepatoprotektory, lipotrooppisia aineita. Asidoosista johtuvia oireita esiintyy laskimonsisäisesti. Munuaisten toimintahäiriö, proteinuria – ACE-estäjät. Hyperurikemialla – allopurinoli. Neutropeniaa – granulosyyttikokoonpanosta stimuloiva tekijä.
- Kirurginen hoito. Potilaat, joilla on vakava kohtalokas maksavaurio, voivat vaatia ortotopoottista elinsiirtoa. Leikkauksen osoitus on maksakirroosi, johon liittyy komplikaatioita, usein kehittymässä kolmannen ja neljännen patologian tyypissä. Joissakin tapauksissa leikkaus on suositeltavaa maksa-adenoomille, joilla on suuri riski muuttaa pahanlaatuiseen kasvaimeen. Munuaissiirto tapahtuu joskus potilailla, joilla on krooninen munuaisten vajaatoiminta.
Ennuste ja ennaltaehkäisy
Hoidon tehokkuus, komplikaatioiden todennäköisyys ja kuolema riippuvat patologian tyypistä. Jotkut glykogeogeenit heikentävät hieman potilaiden elämänlaatua, korvataan kasvaessaan, muut – ei hoidettavissa ja väistämättä päätyä kuolemaan. Vähentää riskiä ottaa vauva glykogeenisaatiosta, riskialttiit parit – joilla on sukututkimus, lapset, joilla on vahvistettu diagnoosi – Geneettinen neuvonta tarvitaan, synnytyksen diagnoosi.