Waardenburgin oireyhtymä
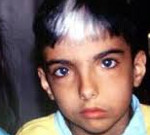
Waardenburgin oireyhtymä on geneettisesti heterogeeninen perinnöllinen sairaus, jolle on ominaista epämuodostumien ja epämuodostumien monimutkaisuus, joka aiheutuu alkion alkamisjakson epämuodostuman rakenteiden rikkomisesta. Oireita tämä ehto on siirretty sivusuunnassa kulmassa molemmissa silmissä, leveä silta ( «kreikkalainen profiilin»), pigmentoitunut iho poikkeavuuksia, hiukset ja iiris kuurous. Oireyhtymän diagnoosi tehdään potilaan nykyisen tilan, audiometristen ja silmälääketieteellisten tutkimusten, perinnöllisen historian ja molekyyligeneettisten tutkimusten perusteella. Tämän sairauden erityistä hoitoa ei ole, terapeuttiset toimenpiteet on tarkoitettu patologian oireiden poistamiseen ja lievittämiseen.
- Vaardenburgin oireyhtymän luokittelu ja syyt
- Waardenburgin oireyhtymän oireet
- Vaardenburgin oireyhtymän diagnosointi ja hoito
Waardenburgin oireyhtymä

Waardenburgin oireyhtymä on perinnöllinen sairaus neurokristopatioiden ryhmästä — patologiat, jotka johtuvat niin sanotun hermosärmän rakenteiden alkionkehityksen rikkomisesta. Ensimmäisen kerran hollantilainen oftalmologi P. Vaardenburg kuvasi vuonna 1951 tämän taudin ominaispiirteen oireiden monimutkaisuutta. Seuraavina vuosina saavutukset genetiikan alalla antavat mahdollisuuden määrittää Vaardenburgin oireyhtymän syyt ja tämän patologian kliinisen kulun vaihtelut. Tämän taudin perinnöllisyysmekanismi on erilainen geneettisen heterogeenisyyden takia — useimmiten on olemassa autosomaalinen hallitseva tyyppi, jolla on epätäydellinen läpäisy, mutta myös autosomaalisia recessive-lajikkeita. Kaikenlaisten Waardenburgin oireyhtymän esiintyminen on noin 1 tapaus 40 000 vastasyntyneelle, eteläisissä Australiassa taajuus on jonkin verran korkeampi, jonka avulla voimme tehdä oletuksen patologian alkuperästä tällä alueella. Lääketieteellisten tilastojen mukaan tämä tauti muodostaa 2-5% kaikista perinnöllisistä kuuroista ja kuuroista. Waardenburgin oireyhtymän seksuaalisella jakautumisella ei ole erityisiä piirteitä, pojat ja tytöt vaikuttavat samalla taajuudella.
Vaardenburgin oireyhtymän luokittelu ja syyt
Tällä hetkellä tunnetaan neljä kliinistä oireyhtymää, joista osa on jaettu alaluokkiin riippuen siitä, mistä mutaatiot ovat johtaneet taudin kehittymiseen. Kaikkien patologisten sairauksien yhteinen syy on hermosärmärakenteiden muodostumisen häiriintyminen alkion aikana — tämä aiheuttaa kasvojen epämuodostumia, pigmenttimuutoksia, kuulo-häiriöitä ja joskus näkyviä. Dysembryogeneesi Waardenburgin oireyhtymään johtuu vikoista geeneissä, joista suurin osa koodaa transkriptioproteiiniproteiineja eli muita geenejä ilmentäviä.
Tyypin 1 oireyhtymä (WS1) on yleisin patologian variantti, jossa on autosomaalinen hallitseva perintötyyppi, mutta eri ekspressiivisyys. Se johtuu toisen kromosomin kohdalla olevista PAX3-geeneistä. Ilmentämisen tuote on transkriptiotekijä, joka oletettavasti ohjaa solujen migraatiota hermosärmässä ja vaikuttaa MITF-geenin ilmentymiseen. Vaardenburgin tyypin 1 oireyhtymän ohella PAX3-geenin mutaatiot johtavat samanlaisen sairauden tyyppi 3: een (WS3) ja sen osallistuminen myös rabdomyosarkooman kehittymiseen on osoitettu.
Wahdenburgin tyypin 2 oireyhtymä (WS2) aiheuttaa noin 20-25% kaikista tämän patologian tapauksista, sillä on vähintään neljä alaluokkaa (a, b, c, d), mutta eniten tutkittuja ovat WS2a ja WS2d. Ensimmäinen johtuu kolmannessa kromosomissa lokalisoidusta MITF-geenistä, se koodaa transkriptiotekijää, joka aktivoi tyrosinaasigeenin, avainentsyymi melanosyyttien kehityksessä. Waardenburg WS2d: n oireyhtymä johtuu 8. kromosomissa sijaitsevan SNAI2-geenin mutaatioista, ja se koodaa myös yhden transkription aktivaatiotekijän. WS2b: n ja WS2c: n aiheuttavien geenien funktiot eivät tällä hetkellä ole selvät, vaan ne sijaitsevat vastaavasti 1. ja 8. kromosomissa.
Waardenburg oireyhtymä 3rd tyyppi (WS3, Klein-Waardenburg syndrooma, joka on nimetty sveitsiläinen lääkäri D. Klein) on vaikein kunnosta. Sen aiheuttaa mutaatio PAX3 geeni (joka on syynä WS1) kuitenkin puutteellinen muoto geenistä potilailla ovat homotsygoottisia tilassa tai hänellä on nonsense-mutaatio.
Waardenburg oireyhtymä tyyppi 4 (WS4, Waardenburgin-Shah oireyhtymä) — on, toisin kuin muut tyypit taudin patologian kanssa peittyvästi periytyvä mekanismi. Siinä on kolme lajiketta (a, b, c), jotka johtuvat erilaisten geenien mutaatioista. EDNRB WS4a aiheuttama mutaatio geenin, joka sijaitsee 13 kromosomiin ja koodaa proteiinia, monimutkainen sekvenssin endoteliini-B. Waardenburg oireyhtymä WS4b molekyyli- patogeneesi on samanlainen kuin edellisessä suoritusmuodossa, koska vika on aiheuttanut EDN3 geeni — se sijaitsee 20. kromosomissa ja koodaa ligandia B-endoteliini. Sitä vastoin WS4c johtuu SOX10-geenin mutaatiosta, joka on edustava laaja joukko sääntelygeenejä. Se sijaitsee 22. kromosomissa.
Waardenburgin oireyhtymän oireet
Waardenburgin oireyhtymän kliininen kuva riippuu pitkälti taudin tyypistä, kun taas kaikentyyppisiä yhteisiä manifestaatioita esiintyy. Siten, minkä tahansa patologian 99%: lla potilaista on sivuttaissiirtymä ulkoreunasta silmän (telekant), kaksi kolmasosaa laajentaminen nenä on tallennettu. Noin puolet kaikista kärsivien henkilöiden oireyhtymä Waardenburg, on valkoinen hiustupsu otsan yläpuolelta, heterophthalmia, synnynnäinen hiustenlähtö leucoderma, kuulonalenema lievempiä lukien kuurous. Tauti on ominaista korkea vaihtelevuus näyttää paitsi eri tyyppejä, mutta myös välillä harjoittajien yhden ja saman poikkeava geeni — saman perheen tai jopa identtisten kaksosten voivat kokea erilaisia oireita.
Waardenburgin tyypin 1 oireyhtymää pidetään klassikkona, ja sitä leimaa lansettelu, ihon pigmentaatio, hiukset ja iiris ja muut tyypilliset kasvojen poikkeavuudet. Minkä tahansa alaluokan WS2 ominaisuus on vain pigmentaation ja kuulon heikkenemisen yksittäisten epämuodostumien esiintyminen — ensimmäisen patologian tyypistä se erotetaan silmän sivusuunnan syrjäyttämisen puuttuessa. Waardenburgin kolmannen tyyppinen oireyhtymä on kliinisesti vakavampi, pigmentaatiohäiriöt, kuurous ja kasvojen epämuodostumat ovat varsin voimakkaita. Lisäksi tämän taudin variantille on ominaista myös lihaskudoksen hypoplasian kehitys, pääasiassa yläraajoissa.
Vaardenburgin tyypin IV oireyhtymän (Wahardenburg-Shah-oireyhtymä) esiintymät sisältävät Hirschsprungin taudin oireet, ruuansulatuskanavan tiettyjen osien kasvullisen innervaation rikkominen. Tälle taudin variantille tyypillisten pigmentaatiohäiriöiden, sensorineaalisen kuulon heikkenemisen ja kuurouden, vatsakivut, ummetus, ilmavaivat ja synnynnäiset paksusuolen laajeneminen ovat megakoloneja. Näiden ilmenemismuotojen vakavuus riippuu paksusuolen osuuden pituudesta, joka ei sisällä submukosaalista hermoplexusta. Lisäksi jotkut tyypin IV oireyhtymät liittyvät hermokuitujen heikentyneeseen myelointiin, lähinnä perifeerisiin — tutkijat erottavat joskus tällaiset tapaukset erillisiksi PCWH-oireyhtymiksi.
Vaardenburgin oireyhtymän diagnosointi ja hoito
Oireyhtymän määrittely tehdään potilaan tietojen perusteella, perinnöllisen historian, silmälääketieteellisten ja audiometristen tutkimusten tutkimiseksi. Diagnoosin lopullinen vahvistus annetaan molekyyligeneettisellä analyysillä. Lisäksi joissakin sairauden muotoja voidaan käyttää, kuten myös muita diagnostisia menetelmiä, kuten elektromyografiaa (at WS3), biopsia paksusuolen seinämän (at WS4). Tutkittaessa, potilaan Waardenburgin oireyhtymä on yleensä havaita tunnusomainen kasvonpiirteet (siirtymä sivusuunnassa kulmat silmät, leveä nenän silta), leucoderma vakavuus vaihtelee, usein nähty valkoinen tuppo hiukset yli hänen otsa. Silmän iiris voi olla erilainen väri (heterochromia) tai lisätä toisen varjon sektorin muodossa.
Audiometriset tutkimukset Waardenburgin oireyhtymässä osoittavat usein kuulon heikkenemisen täydellisen kuurouden loppuun asti. Yleensä kuulovamma tämän taudin kanssa ei yleensä ole edistynyt. Harvinaisissa tapauksissa, Waardenburgin oireyhtymä voi liittyä synnynnäinen sydänvika, suulakihalkioita ja muut vakavat sairaudet 3. sairauden tyyppi on määritetty hypoplasia lihakset yläraajojen. Epäillään WS4 tuotteiden sijaan radiografia paksusuolen, läsnä ollessa laajennetun osien suolen koepaloja suoritetaan seinät niiden alueella — tapauksessa Waardenburgin oireyhtymä tyyppi 4 on havaittu ilman hermopunoksen. Geneettinen geenitekijä voi suorittaa tämän taudin lähes minkä tahansa muodon molekyyligeneettisen diagnoosin sekvensoimalla siihen liittyvät geenit.
Tähän tautiin ei ole olemassa erityistä hoitoa, vaan vain oireenmukaista hoitoa sekä potilaiden elämänlaadun parantamiseen tähtääviä toimia. Kuulon menetys ja kuurous kanssa Waardenburgin oireyhtymä voi kompensoida sisäkorvaistutteeseen laite ja se tulisi tehdä mahdollisimman varhain normaalille kehitykselle lapsen puheen. Näiden tai muiden silmäsairauksien kehittymisen yhteydessä saattaa olla tarpeen suorittaa korjaus silmälasien tai piilolinssien muodossa. Tyypin III oireyhtymään tyypillisten lihasten hypoplasia saattaa vähentyä säännöllisin harjoituksin fysioterapian ja fysioterapian käytön yhteydessä. Ruoansulatuskanavan vaurio WS4: llä eliminoidaan kirurgisesti — poistamalla patologisesti muutetut suoliston alueet.
Waardenburgin oireyhtymän ennuste ja ennaltaehkäisy
Useimmissa tapauksissa oireyhtymän ennuste on suotuisa, koska tämän sairauden kehittymisvaikeudet eivät useinkaan uhkaa potilaan elämää eikä heillä ole taipumusta edetä. Harvinaisissa tapauksissa tämän patologian (sydämen vajaatoiminta, suolistovaikutus, ojentajälki, hydrokefalus) lisäilmiöiden poikkeavuudet voivat hieman pahentaa ennusteita. Vaardenburgin oireyhtymään vaikuttavien potilaiden elämänlaadun vähentämisen tärkein tekijä on kuurous ja kuurous, jotka myöhäisessä diagnoosissa voivat johtaa puheen kehittymisen häiriöihin. Usein silmien ja ihon pigmentaation heikkenemisen vuoksi tämän patologian omaavilla ihmisillä on auringonpoltto ja lisääntynyt herkkyys valolle, joka on poistettu aurinkolasien avulla ja käyttämällä erityisiä voiteita.