Bullous epidermolysis
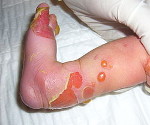
Epidermolysis bullosa — joukko perinnöllisiä sairauksia, joille on tunnusomaista valo ihon heikkoutta, joten toinen nimi näiden sairauksien — «mehanobulleznaya tauti.» Tärkein oire on kehittää pinnalla ihon rakkuloita kanssa seroosiset sisältöä, ja sitten niiden tilalle on pitkät parantumattomissa eroosiota. Vikatyypin rakkulaihotauti suoritetaan käyttämällä immunohistologisilla ja geneettisiä tekniikoita sekä perusteella tarkastelu potilastiedot ja tarkastus sen perinnöllinen historiaa. Spesifistä hoitoa ei ole olemassa, vaan oikea ja kattava oireenmukainen hoito voi joissakin tapauksissa merkittävästi parantaa potilaan vointia.
- Syyt bullous epidermolysis
- Bullous epidermolyysi luokitus
- Bullous epidermolysis oireet
- Bullous epidermolyysi diagnosointi
- Bullous epidermolyysihoito
- Bullouksen epidermolyysipotilaiden ennustaminen
Bullous epidermolysis
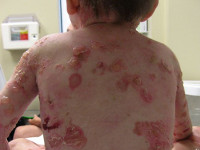
Epidermolysis bullosa — heterogeeninen ryhmä perinnöllisiä ihosairauksia, tunnettu siitä rakkuloita ja eroosiot, vasteena hieman mekaanisesti. Ensimmäistä kertaa kyseistä sanaa käytetään vuonna 1886 Saksan ihotautilääkäri Henry Kobnerom, lisätutkimukset ovat osoittaneet, että on olemassa monia lajikkeita tämän taudin. Geneettinen tutkimus rakkulaihotauti ovat osoittaneet, että sitä voidaan periytyvät autosomaalinen peittyvästi periytyvä ja autosomaalinen dominantti, mutaatiot liittyvät häntä enemmän kuin 10 geenit. On merkittäviä eroja kliinisen erityyppisten taudin ilmaantuvuus vaihtelee 1: 30000-1: 1000000.
Bullous epidermolyysiin liittyvien häiriöiden patogeneesi on pitkään ollut huonosti ymmärretty. Läpimurto tähän suuntaan tapahtui ottamalla käyttöön elektronimikroskopian lääketieteellinen käytäntö, joka auttoi visualisoimaan vaikutuksen kohteena olevan ihokudoksen ultrastruktuurin. Seuraava tärkeä vaihe bullous epidermolyysiä tutkittaessa oli immunohistologisten tutkimusten (immunofluoresenssin) löytäminen. Tällä hetkellä näillä menetelmillä on ratkaiseva merkitys näiden tautien diagnosoinnissa, jolloin saadaan vain geneettinen analyysi tarkkuuteen. Ottaen huomioon, että bulloosin epidermolyysiä koskevia menetelmiä on jatkuvasti parannettu, on muutettu ja luokiteltu tämän sairausryhmän muodot.
Syyt bullous epidermolysis
Bullous epidermolyysi etiologia ei ole sama eri tautityypeissä, mikä joissakin tapauksissa vaikeuttaa diagnoosin riittävän hyvin. Yksinkertainen epidermolysis bullosa mutaatioiden aiheuttama KRT5 ja KRT14 geenit, kuitenkin, mukaan lääkärin geneetikot, nämä geenit rikkoo rakenne selittää ainoastaan 75% tapauksista taudin tämä tyyppi. Tässä tapauksessa ihon ihon oletetaan häiriintyvän entsyymi-inhibiittorijärjestelmässä, ja jotkin proteiinit tulevat hyökkäyksen kohteiksi. Yksinkertaisella epidermolysis bullosa se voi olla tyvikalvon proteiineja (alfa6 integriini-beta4) ja desmosomiproteiinien tyvikerroksen orvaskeden — desmoplakin, plakofillin-1. Tämän seurauksena, kun mekaaninen voima vapautetaan entsyymejä, jotka hajottavat näitä proteiineja, saattaen tuhoaminen orvaskeden ja sytolyysi rakenne, jolloin muodostuu kuplia.
Syynä kehon toisen patologian muodon — rajan bullous epidermolysis — ovat mutaatioita geenejä LAMB3, LAMA3 ja jotkut muut. Suurin osa näistä mutaatioista periytyy autosomisen recessive -mekanismin avulla, epäsymmetrisen entsyymijärjestelmän hyökkäyksen kohteena ovat proteiinit, kuten kollageenityyppi 17 ja laminini 332. Nämä proteiinit osallistuvat epidermiksen alemman kerroksen normaalin rakenteen ylläpitämiseen, joten niiden vaurioituminen aiheuttaa luontaisen epubenolyysirajoituksen luonteenomaisia kliinisiä oireita. Kuplien ja eroosion helppoon muodostumiseen liittyy myös ihon lisääntynyt hauraus ja vaikeampi kuristus.
Dystrofinen epidermolysis bullosa tyyppi johtuu mutaatioista COL7A1 geeni, joka voidaan periytyvät autosomaalinen dominantti ja peittyvästi periytyvä mekanismeja. Kohdeproteiinin toimii siten kollageenin tyyppi-7, joka on vastuussa vakauden ihon muita rakenteita sidekudosten kuituja. Vähentämällä tämän proteiinin kudoksissa ihon johtaa helposti kehittämiseen ihottumaa, rakkuloita ja eroosiot, ja usein liittyy häiriöt muihin elimiin. Erityisesti dystrofiset rakkulaihotauti johtaa usein kehittää yhteisiä kontraktuuria, tappio kaappaa limakalvojen hengitysteiden ja ruoansulatuskanavan järjestelmiä. Siitä arvet, jotka jäävät jäljelle, kun parantavaa eroosiot usein syntyy syöpäsairauksia.
Yleensä yhteinen patogeneesi rakkulaihotauti voidaan vähentää rikkoo toiminnan tiettyjen entsyymien ihon kudoksia. Tämän seurauksena, se tuhosi tiettyjä keskeisiä rakenteellisia proteiineja orvaskeden, verinahan tai tyvikalvon, joka antaa viestintä solujen välillä ja johtaa kuplien muodostumista mekaanisen vaikutuksen jopa pieni voima. Epidermolysis bullosa tyypit eroavat toisistaan paikallinen kuplia, näkymät mutaatio, joka johti taudin, ja proteiini lajit, joka tuli kohde hyökkäys entsyymejä.
Bullous epidermolyysi luokitus
Tällä hetkellä on kymmeniä bulloos epidermolyssia, joita on vaikea luokitella tietyissä ryhmissä. Ongelman monimutkaisempi on se, että lähes vuosisadan ja puoli tämän patologian tutkimiseen on tehty toistuvia yrityksiä jakaa se tiettyihin tyyppeihin käyttämällä nykyaikaisinta tietoa. Lopulta tämä johti sekaannukseen, jopa tieteellisessä kirjallisuudessa, erilaisia vaihtoehtoja bulloosin epidermolyysierojen erottamiseksi lajikkeelle. Tämän ehton nykyaikaisimmassa luokassa dermatologiassa on neljää tautityyppiä, jotka puolestaan jaetaan useisiin alatyyppeihin:
- Yksinkertainen rakkulaihotauti — on 12 alatyyppejä, joista yleisimmät ovat Weber-Cockaynen oireyhtymiä, Kobnera, Dowling-Meara. Voidaan periä autosomaalisena dominanttina, ja recessively esiintyminen on 1: 100,000. Yksinkertainen epidermolysis bullosa tunnettu muodostamalla säären tai, harvoin, subepidermaalinen rakkuloita, koska tämä sairaus vaikuttaa orvaskeden proteiineja.
- Rajoitettu bullous epidermolyysi on jaettu kahteen alatyyppiin, joista yhdellä on 6 erillistä kliinistä muotoa. Tämän taudin vakavin muoto on Herlitz-alatyyppi, jolla on erittäin korkea kuolleisuusaste. Reunan bulloosin epidermolyysi esiintyy noin 1: 500000, läpipainopakkausten muodostuminen tapahtuu valolevyn tasolla, mikä antoi sille nimen «rajat».
- Dystrofinen bulloos epidermolysis — on kaksi alatyyppiä, jotka jakautuvat tämän patologian perimismekanismin (hallitsevat ja recessive altatyypit) mukaisesti. Tässä tapauksessa hallitsevan variantin esiintyminen on jonkin verran korkeampi (3: 1 000 000 verrattuna 1: 500 000: aan dystrofisen bulloosin epidermolyysiherkässä muodossa). Recessive-lajikkeella on myös useita kliinisiä muotoja, joista vakavin on Allopo-Siemens-alatyyppi. Tällä taudin variantilla potilaat kehittävät syviä eroosiota, jättäen arvet taakse, nivelet voivat olla supistuneet ja limakalvot voivat vaurioitua. Ristikkäiden muodostuminen esiintyy dermis-papillon kerroksessa, mikä aiheuttaa arpia ja pidentää eroosiota.
- Kindler-oireyhtymä tai sekalaisen bulloosin epidermolyysi on yksi tämän patologian harvinaisimmista ja huonosti tutkituista muodoista. Ominaisuus, joka erottelee tämän muodon erillisessä tyypissä, on kuplien muodostuminen kaikissa ihon kerroksissa — ihon kuorikerroksessa kevyessä levyssä. Tällä hetkellä määritellään vain proteiineja, jotka toimivat entsyymien kohteena sekalaisen bulloosin epidermolyysi — kyndlin-1 kanssa.
Tällaisen bulloosin epidermolyysin kaikkien kliinisten muotojen tällainen erottelu on nyt yleisesti hyväksytty. Mutta jopa yhden tyypin sisällä on taudin kliinisiä oireita, jotka vaikeuttavat diagnoosia ja usein vaikuttavat patologian ennusteisiin. Siksi tähän mennessä on pyritty etsimään rakenteellisempaa ja hyväksyttävää bulloosin epidermolyysiä.
Bullous epidermolysis oireet
Eri tyyppisten bullous epidermolyysi ilmenee yhdestä — rakkuloiden kehittymisestä ja eroosiosta johtuen ihon mekaanisista vaikutuksista. Näiden muutosten ilmaisuaste, lokalisointi, olemassaolon aika ja paranemisen tulokset poikkeavat toisistaan. Yksinkertaisen bulloosin epidermolyysi (Weber-Kokkein-alatyyppi) lokalisoidussa muodossa leesiot sijaitsevat vain tiettyyn kehon osaan (kädet, jalat). Alkuvaiheessa rakkuloiden ulkonäön laajempi alue on mahdollinen, mutta niiden vakavuus vähenee iän myötä. Päinvastoin, Dowling-Mearen yleistynyt alatyyppi on luonteenomaista pienten vesikulaaristen purkausten kehittyminen suuren kehon alueen yli. Tämäntyyppinen bullous epidermolyysi ilmenee varhaislapsuudesta ja voi aiheuttaa lapsen kuoleman, rakkuloiden erottelun tulos voi olla hyperkeratoosi, ihon pigmentaatiohäiriöt,
Bullous epidermolyysin rajat ylittävä muoto etenee paljon voimakkaammin, erityisesti Herlitzin ns. Lethal-alatyypistä. Tässä tapauksessa ihon epävakaisuus lisääntyy, suuri määrä rakkuloita, eroosiota, kasvojen ja selän usein on symmetrisiä rakeita. Vaikuttaa suun limakalvoihin, emalian hypoplasia ja sen aiheuttamat vakavat karieksit. Tällainen rajallinen bullous epidermolysis aiheuttaa usein kuoleman ensimmäisinä elämänvuoroina. Aikuisikään sairastuneissa potilailla muodostuu niveltymiä, munuaisvaurioita, kynsien menetystä. Reunan bulloosin epidermolyysiin kevyempää atrofista muotoa leimataan myös laajoilla ihottumilla, jonka jälkeen atrofiset laastarit ja arvet muodostuvat.
Dystrofinen rakkulaihotauti on lähes aina yleistynyt ja vaikuttaa suureen osaan kehon. Määräävässä asemassa sairauden variantin koko on enemmän hyvänlaatuinen tietenkin kuplien ja niiden ratkaiseminen on hidas, mutta useimmat potilaat lopulta menettävät kynnet. Eroosion parantumisen jälkeen näkyvät arvet muodostavat ihon pinnalle. Peittyvästi dystrofinen rakkulaihotauti vaihtoehto, varsinkin sen raskasta yleistynyt alatyyppiä esiintyy paljon vaikeampaa: lisäksi ihottumaa potilailla usein kirjataan psevdosindaktilii, laaja arpia, menetys nauloja. On menetys luita, sivuston parantunut arpi vuosien voi kehittyä okasolusyöpä. Ongelmana on myös Allopo-Siemens-alatyypin stabiilius terapeuttisiin toimenpiteisiin.
Komplikaatiot tahansa epidermolysis bullosa vähentää sokki (joilla on laajoja vaurioita), liittyä toisen tartunnan ja sepsis aiheuttama se, kuivattu potilailla. Useimmissa tapauksissa terapeuttiset toimenpiteet suoritetaan vain näiden olosuhteiden estämiseksi. Komplikaatioiden todennäköisyys on suurempi, mitä suurempi on kehon alue, jolla patologiset fokaalit vievät, ja niiden luonteeltaan tuhoisammat (kudotut kuplat, eroosiot, haavaumat).
Bullous epidermolyysi diagnosointi
Tällä hetkellä bulloosin epidermolyysi diagnosoidaan tutkimalla potilaan ihoa, suorittamalla immunohistologisia tutkimuksia ja geneettisiä analyysejä, joissakin tapauksissa perinnöllisen historian tutkimusta. Ihon tutkimista varten erikoislääkäri voi myös tehdä diagnostisia testejä — mekaanisesti vaikuttaa potilaan ihoon ja arvioida tulokset jonkin ajan kuluttua. Bullous epidermolyysiin liittyvien kuplien tai eroosion kehittyminen tällä sivustolla puhuu tämän taudin läsnäolon puolesta. Diagnoosin seuraavissa vaiheissa tehdään patologisen lomakkeen tarkempi määrittely.
Immunofluoresenssianalyysi epidermolysis bullosa suorittaa käyttäen mono- tai polyklonaalisia vasta-aineita, joilla on affiniteetti pääproteiineja iho, valon levy ja ylempien kerrosten dermis. Tämän avulla on mahdollista arvioida määrä proteiinia, joka puolestaan puhuu entsyymiaktiivisuuden kudoksissa. Proteiinin määrän vähentäminen osoittaa sen alhaisen erittymisen tai nopeutetun tuhoutumisen. Pitoisuuden pienentämiseksi avaimen proteiinien tietyillä alueilla tason määrittämiseksi kehityksen kuplien varhaisessa vaiheessa, mikä auttaa jo suurella todennäköisyydellä määrittää tyypin epidermolysis bullosa. Kohta diagnosointiin tämä ehto asettaa geneettisen analyysin suoralla sekvensoinnilla geenejä, jotka liittyvät tietyn tyyppisen sairauden.
Yksinkertaistavat diagnoosi tämän sairauden mahdollistaa tutkimuksen perinnöllinen sairaushistoria potilaan, joilla voit tunnistaa hänen verensä sukulaisten kanssa sama ongelma. Lisäksi, jos joku perhe on rakkulaihotauti, on järkevää tehdä synnytystä geneettisen diagnoosin, joka tunnistaa läsnäolo tämän taudin alkuvaiheessa sikiön kehitykseen. Erotusdiagnoosissa suoritetaan totta pemfigus, joitakin muotoja rakkulainen pemphigoid, hankittu epidermolysis bullosa (joka ei ole perinnöllinen, ja autoimmuunisairaus).
Bullous epidermolyysihoito
Tämän taudin erityistä hoitoa ei ole, kaikki terapeuttiset menetelmät vähenevät komplikaatioiden kehittymisen estämiseksi ja vesikkelien vakavuuden ja eroosiota vähentäen. Jos kyseessä on vaikea bullous epidermolyysi, prednisoloni on määrätty. Ulkopuolisista terapeuttisista manipulaatioista rakkuloiden aseptinen avautuminen, niiden kansien hoito antiseptisilla aineilla, määrää heliomysiinivoiteen. Kastelujen levittämistä on tehtävä hyvin huolellisesti, koska siteiden paine voi aiheuttaa uusia rakkuloita. Komplikaatioiden (sokki, sepsis) esiintyminen, oireenmukainen hoito anti-shokkilla ja antibiooteilla. Ehkäisevällä tarkoituksella on mahdollista säteilyttää ihoa ultraviolettisäteillä.
Moderni genetiikka ja monet muut lääketieteen alat jatkavat laaja-alaisia tutkimuksia bulloos epidermolysis löytää tehokkaampia hoitomenetelmiä. Tärkeimmistä tekniikoista ja menetelmistä lupaavimpia ovat menetelmät, joissa käytetään kantasoluja, proteiinia ja geeniterapiaa. Toistaiseksi yksikään menetelmä ei kuitenkaan ole ylittänyt eläinkokeita, joten sonniepidermolyysi on parhaillaan parantumatonta tautia.
Bullouksen epidermolyysipotilaiden ennustaminen
Ennuste rakkulaihotauti usein epävarmaa, sillä se riippuu monista tekijöistä, ja olosuhteet — sairauden tyypin, läsnäolo tai puuttuminen liittyviä häiriöitä potilaalla, hänen elämäntapansa. Esimerkiksi yksinkertainen paikallinen alatyypin epidermolysis on yleensä hyvänlaatuinen kurssin ja harvoin uhkaa elämää potilaalle. Kun taas alatyyppi Allopo-Siemens on erittäin korkea kuolleisuus — samoin kuin iho-oireita, ja koska pitkäaikaisia komplikaatioita, kuten munuaisvaurioita ja ruoansulatuskanavassa sekä kehitystä okasolusyöpä ihosyöpä. Potilaat, joilla tämä ongelma täytyy pitää hyvää huolta ihon, älä unohda antiseptinen hoito eroosion ja muiden vaurioiden välttämiseksi toimintaa traumaattinen urheilua ja muita vastaavia toimintoja.