Pompe-tauti
Pompen tauti — harvinainen perinnöllinen poikkeavuus, muoto lysosomaalisiin tunnusomaista heikentynyt glykogeeni pilkkomalla prosesseja hermo- ja lihassoluissa (luustolihas, sydänlihas). Sairauden oireita on varsin vaihteleva aika sen ilmenemismuotoja ja vakavuuden eri potilailla, perinteisesti havaittu progressiivinen lihasheikkous joissakin muodoissa — kardiomegaliaa ja laajentuneet kardiomyopatia. Pompen tauti diagnostiikka suoritetaan perusteella suvussa, histologisia ja histokemiallisia tutkimus lihaskudoksen veren biokemiallisia analyysi, ja geenitutkimuksen. Käsittely tällä hetkellä voidaan valmistaa käyttämällä entsyymiä-korvaushoitoa, mutta tehoa tämän menettelyn vaihtelee potilaiden välillä.
- Pompe-taudin syyt
- Pompe-taudin luokittelu
- Pompe-taudin oireet
- Pompe-taudin diagnosointi
- Pompe-taudin hoito ja ennuste
Pompe-tauti
Pompen tauti (glykogenoosi tyyppi 2 vajaatoiminta hapanta alfa-glukosidaasia) — perinnöllinen sairaus, jossa vastaisesti glykogeenin metabolian prosessien vahinkoa hermo- ja lihaskudokseen. Ensimmäinen kerta, kun hollantilainen tiedemies I. Pompe kuvasi sitä vuonna 1932, sen jälkeen yli 50 patologian tapausta on virallisesti kirjattu. Pompen tauti yhtä suurella todennäköisyydellä vaikuttaa sekä miehet ja naiset, ilmaantuvuus vaihtelee välillä 1: 60000 (kypsässä muodossa) ja 1: 140000 (aikaisin tai lapsuusajan muodossa). Se on yksi harvoista lysosomaalisiin, johon se on kehittänyt tehokkaan erityistä hoitoa hyväksytty Yhdysvalloissa vuonna 2006 ja Venäjällä — vuonna 2013. Kuitenkin arvo syy Pompen taudin hoidosta on erittäin suuri ja sen määrä on useita satoja tuhansia dollareita vuodessa.
Pompe-taudin syyt
Pompe-tauti on klassinen glykogeenisaatio, jossa glykogeenipitoisuudet muodostuvat luustolihaksen kudoksissa, sydänlihaksessa ja osittain hermojärjestelmässä, koska sen pilkkoutuminen on mahdotonta. Tämä tapahtuu 17-kromosomin kohdalla olevan GAA-geenin mutaation seurauksena — se koodaa happo-alfa-1,4-glukosidaasin tai maltaasin sekvenssiä. Tämä on yksi lysosomien keskeisistä entsyymeistä, joka liittyy glykogeenimolekyylin pilkkoutumiseen yksinkertaisemmiksi segmentteiksi, mikä lopulta hajoaa glukoosin sisään tulevan solun energian metaboliaan. Koska glykogeeni on tärkeä energiavarasto rakenteille, kuten luurankolihaksille, sydänlihakselle, maksalle ja hermokudokselle, Pompe-taudin oireet vähenevät näiden elinten patologisiin muutoksiin.
Tällaisten muutosten seurauksena syntyy ensin energiapuutteita soluissa — glukoosin kudoksen tarpeet katetaan vain sen saannosta verestä. Lisäksi Pompe-taudin lysosomeissa glykogeeni alkaa kertyä muodostaen suuria inkluusioita vakuolien muodossa, mikä edelleen johtaa dystrofiaan ja solujen vaurioitumiseen. GAA-geenin viallisten muunnosten perintö tapahtuu autosomaalisessa recessiivisessa muodossa. Useiden taudinmuotojen esiintyminen johtuu oletettavasti erilaisten edellä mainitun geenin mutaatioista. Ehkäpä tietyillä vikoilla ei ole täydellistä katoamista vaan vain happaman alfa-1,4-glukosidaasin aktiivisuuden väheneminen, mikä johtaa Pompe-taudin myöhempiin kehittymiseen ja taudin hidas etenemiseen. Patologian muodon määrittämisellä on tärkeä rooli ennusteidensa ja hoitojärjestelmänsä muotoilussa.
Pompe-taudin luokittelu
Tähän mennessä asiantuntijat tunnistavat useita Pompe-taudin merkittäviä muotoja, joista tärkein ero on taudin puhkeamisen ajoitus ja oireiden vakavuus. Useimmissa tapauksissa tyypin 2 glykogeenisaatio kokee lastenlääkäreitä, mutta on sellainen sairaus, joka havaitaan aikuisilla.
- Varhainen infantilainen muoto — tämän tyyppistä Pompe-tautia pidetään vakavimpana. Se löytyy jo ensimmäisten elämänkuukausien aikana, myopatian, maksavaurion (hepatomegalia) ja sydämen (kardiomyopatian) oireet kehittyvät nopeasti. Tyypillisesti potilaat, joilla on varhaisen infantilainen Pompe-sairauden muoto, kuolevat sydän- tai hengitysvajauksista ennen ikävuotta.
- Myöhäinen lapsenmuoto — ensimmäiset oireet esiintyvät 1-3 vuoden iässä, ja heidän etenemisnopeutensa on myös paljon hitaampaa. Tämäntyyppisellä Pompe-taudilla sydänlihaksen vauriot ovat voimakkaimpia, kuolema tulee nuoruusiässä sydämen vajaatoiminnasta.
- Pompe-sairauden nuorten muoto kehittyy 6-10-vuotiaana. Aivan kuten edellisessä versiossa, sydän on taudin tärkein kohderyhmä, kasvava sydämen vajaatoiminta kuolee 20 vuoteen.
- Pompe-taudin aikuinen muoto — oireiden ilmaantuminen tapahtuu 20-40 vuodessa. Varsinainen oire on hitaasti etenevä myopatia, maksavaurioita on lähes koskaan kirjattu, joissakin tapauksissa pienet sydänlihasten rikkomukset ovat mahdollisia. Tällaisessa Pompe-taudissa potilaat monissa tapauksissa selviävät vanhuksille, mutta vain joskus aikaisempi tappava lopputulos on mahdollinen hengitys- tai sydämen vajaatoiminnan takia.
Modernin genetiikan menetelmillä tällä hetkellä ei ole määritetty suhteita yksittäisten GAA-geenimutaatioiden ja Pompe-taudinmuotojen välillä. Todennäköisesti ilmiöiden tällaisen vaihtelun syy on aivan toisessa — kirjallisuudessa kuvataan taudin perhetapauksia, kun sukulaiset rekisteröivät erilaisia patologisia muotoja. Tietyyppisen Pompe-taudin kehittymiseen johtavien sääntöjen tutkiminen suurenee huomattavasti tämän oireyhtymän suhteellisen vähäisyyden vuoksi.
Pompe-taudin oireet
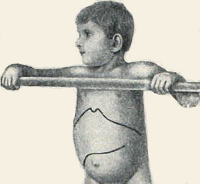
Pompe-taudin puhkeaminen on melko erilainen taudin eri muodoissa. Varhainen lapsen tyyppi on ominaista voimakas lihaskuntoinen heikkous, moottoritoiminnan heikkeneminen, kyyneleet. Pediatriassa, kun potilasta tutkitaan, psykomotorisen kehityksen viivästyminen, erilainen maksa-asteiden suureneminen, palpataatio paljastaa joskus lihasten hypertrofiaa, joka on kuitenkin melko heikko. Pompeen taudin kehittymisen myötä ruokkimisongelmia ilmenee imukuppion heikkouden vuoksi, dysfagia paljastuu ja kaiken tämän seurauksena hypotrofia. Lisääntyvä kardiomyopatia ja heikkous hengityslihaksissa johtavat lopulta lapsen kuolemaan.
Pompe-tautien myöhäiset lapsi- ja nuorisomuutokset esiintyvät lähes identtisesti, mutta patologian oireiden alkamisajankohta poikkeaa toisistaan. Yleensä lihasheikkous, kardiomyopatian merkit paljastuvat. Ajan myötä luustolihaksen voimakasta dystrofiaa, kardiomegalia alkaa muodostua, taustalla maksa ja perna alkavat lisääntyä. Näiden Pompe-tautien muodon kesto on noin 10-12 vuotta, minkä jälkeen hoidon puuttuessa on dekaanieniivisen sydämen vajaatoiminnan seurauksena kohtalokas lopputulos. Epäsuora oire on suuri taudinpurkaus, jossa on keuhkoihin liittyviä komplikaatioita, yöllinen apnea, päänsärkyä aamulla.
Pompe-taudin aikuisille on tyypillistä myöhäinen puhkeaminen (ensimmäiset ilmenemismuodot ilmenevät 20-40 vuodessa). Kliinisesti ennen kaikkea distaalinen myopatia, joka ilmenee ääripäiden lihasten heikkoudesta, havaitaan. Ajan myötä selkärangan kaarevuus (skolioosi, lordoosi) johtuen lihaskotelon ympäröivän selkärangan heikkoudesta. Joissakin tapauksissa on havaittu hitaasti kasvavia sydämen vajaatoiminnan merkkejä, mutta tällaisen Pompe-taudin kanssa sydänvaurioita ei aina tapahdu. Aikuisen patologian muoto kestää useita vuosikymmeniä, joten joissakin tapauksissa potilaat elää vanhuksille, joilla on hyväksyttävä elämänlaatu ja ilman erityistä hoitoa.
Pompe-taudin diagnosointi
Tunnistaminen Pompen tauti voidaan tehdä lukuisia kliinisillä tekniikoilla — biokemiallinen verikoe, Lihasbiopsia tutkimukset, viljelmät fibroblastien tai leukosyyttien potilas, tavanomainen tutkimus (tutkimus, EKG, sydämen ultraäänitutkimus). Tutkittaessa usein paljastaa heikkous ja rappeutumista lihaksia, jossa osallistuminen patologisen prosessin sisäelinten — ja maksa- splenomegaly. EKG on tallennettu PQ-aika lyhentäminen, suurempi kompleksinen QRS, koska koon kasvu infarktin. Tästä syystä, lisäämällä ventrikulaarisen repolarisaation kestoa vaihe, joka ilmenee aaltoinversio T. sonografinen tarkastelu sydämen osoittaa jyrkkä nousu sen koon vuoksi merkittävää paksuuntumista kammion seinät. Aikuisten muodossa Pompen tauti infarktin edellä muutoksia ei voida havaita.
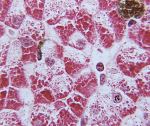
Veren biokemiallinen analyysi voi havaita taudin spesifiset ja epäsuorat oireet. Erityinen on selvittää aktiivisuuden hapanta alfa-1,4-glukosidaasin veriplasmassa, joka Pompen tauti on vähentynyt voimakkaasti. Epäsuora indikaatio patologian esiintymisestä on kreatiinifosfokinaasin voimakas lisääntyminen, joka johtuu lihaskudoksen vaurioista. Lihaksen biopsian histologinen tutkimus paljastaa myosyytteissä lukuisia glykogeenin inkluusioita, usein antaen heille «vaahtosolujen» ilmestymisen. Histokemiallista tutkimus Pompen tautia paljastaa jyrkkä lasku aktiivisuuden hapanta alfa-1,4-glukosidaasin aktiivisuus lihaskudoksen, fibroblasteissa ja leukosyyteissä.
Perinnöllisyyslääkärin voidaan suorittaa geneettinen määrittely Pompen tauti — se on valmistettu suoralla sekvensoinnilla GAA geenisekvenssin tunnistamiseksi viallisten osuuksien. Lisäksi se voi auttaa sairauden diagnosoinnissa ja perinnöllisen anamneesin tekemisessä. Geneettinen diagnoosi Pompen tauti liittyy sekvensointi GAA ja fenotyypiltään terve sukulaisia potilaan tunnistamiseksi kantajia epänormaali geenin.
Pompe-taudin hoito ja ennuste
Tähän mennessä Pompe-taudin ainoa erityinen hoitomenetelmä on entsyymikorvaushoito hapon alfa-1,4-glukosidaasin puutteen täyttämiseksi. Voit tehdä tämän käyttämällä Yhdysvaltojen tuottaman lääkkeen alfa-alglukosidaasia. Tämän hoidon kustannukset ovat erittäin suuret (vuotuinen korko on 100-400 tuhatta dollaria), mutta sen tehokkuus ei ole sama eri potilaille. Tällä hetkellä ei ole muita tapoja hoitaa Pompe-tautia. Ilman hoitoa lapsille ja nuorille sairauksille ennuste on epäsuotuisa, aikuisen tyyppinen — epävarma. Prophylaxis on mahdollista vain Pompe-taudin kuljettamisen ajallaan (jos patologian esiintyminen veren sukulaisissa on) ja myöhemmän geneettisen prenataalisen diagnoosin.