Pigmentoitunut retiniitti
Pigmentti-retiniitti on geneettisesti heterogeeninen perinnöllinen sairaus, jolle on ominaista verkkokalvon pigmenttiepiteelin heikentynyt toiminta erilaisten häiriöiden kehittymisen myötä. Oireiden esiintyminen ja oireiden vakavuus riippuvat patologian muodosta, useimmiten näkökentän vakavuuden ja kaventumisen, skoton kehittymisen ja tumman sopeutumisen rikkomisen vähentyessä, sokeus voi ilmetä. Diagnoosi retinitis pigmentosa tehdään silmälääketieteellisten tutkimusten (fundus, electroretinography and electrooculography) tutkimusten perusteella, molekyylien geneettiset analyysit. Tämän sairauden erityistä hoitoa kehitetään parhaillaan (geeniterapia, kantasolujen käyttöä) kliinisessä käytännössä, tukihoitoa.
- Pigmentin retiniitin syyt ja luokittelu
- Retinitis pigmentosan oireet
- Diagnoosi retinitis pigmentosa
- Pigmentin retiniitin hoito ja ennuste
Pigmentoitunut retiniitti
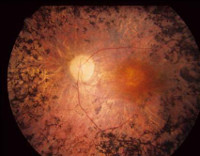
Pigmentin retiniitti (retinitis pigmentosa retinitis) on perinnöllinen degeneratiivinen verkkokalvon sairaus, jolle on tunnusomaista vakavan näkökyvyn kehittyminen täydelliseen sokeuteen saakka. Tämä sairaus, joka on yksi aikakauden visuaalisen näköhäiriön syistä, on tunnettu antiikin ajoista lähtien, mutta hollantilainen oftalmologi F. Donders ehdotti vuonna 1857 termiä «pigmentti retinitis». Oftalmologian ja genetiikan kehittymisen myötä oli mahdollista saada selville, että tämä tila on koko joukko verkkokalvon sairauksia, joilla on erilainen etiologia, mutta samanlainen patogeneesi. Tällä hetkellä on olemassa useita kymmeniä geenejä ja satoja muunnelmia muunnoksista, jotka voivat johtaa tähän sairauteen. Retinitis pigmentosa -perheen mekanismi voi myös olla erilainen — kuvataan autosomaalinen dominantti, autosomaalinen recessiivinen ja liittynyt X-kromosomipatologiaan. Jälkimmäisistä, myös recessive (vain miehet ovat sairas) ja hallitseva (vaikuttavat molempien sukupuolten kasvot) on erotettu. Retinitis pigmentosan keskimääräinen arvo on suuruusluokkaa 1: 5000, mutta taudin muodot ovat sekä suurempia että harvemmin esiintyviä. Lääketieteen tilastojen mukaan vähintään 100-120 miljoonaa ihmistä on geneettisiä vikoja (mukaan lukien oireeton kuljetus).
Pigmentin retiniitin syyt ja luokittelu
Pigmentin retiniitin etiologia on hyvin monimuotoinen johtuen tämän taudin geneettisestä heterogeenisuudesta. Nykyään eritellään lukuisia tämän sairauden muotoja, jotka johtuvat erilaisten geenien mutaatioista. Yleensä retinitis pigmentosan syy ovat fotoreseptoreihin ja pigmenttiepiteeliin liittyvät metaboliset häiriöt, mikä johtaa myrkyllisten sivutuotteiden kertymiseen verkkokalvoon. Taudin nykyaikainen luokittelu perustuu geneettisen häiriön perinnöllisen siirron mekanismiin. Tämän kriteerin mukaan neljä pigmentti-retiniitin ryhmää määritellään.
Retinitis pigmentosa kanssa autosomaalinen dominantti perintö — on yleisin patologia suoritusmuodossa erilaisia tietoja on 70-90% kaikista tapauksista. Syy tähän muodossa verkkokalvon abiotrophy voivat toimia geenin mutaatio RP1 (kromosomi 8), PRPH2 (kromosomi 6), RP9 ja IMPDH1 (kromosomissa 7), ja monet muut. Kaikki nämä geenit koodaavat proteiineja, jotka osallistuvat pigmenttiepiteelin aineenvaihduntaan, joten niiden rakenteessa olevat häiriöt johtavat erilaisiin visuaalisiin häiriöihin. Autosomaalinen hallitseva retinitis pigmentosa, huolimatta suuresta esiintymisestä, hyvin lieviä sairauksia, hidastaa etenemistä, että riittävä ylläpitohoito joissakin tapauksissa voidaan merkittävästi hidastaa tai jopa estää kehitystä sokeutta.
Retinitis pigmentosa kanssa peittyvästi periytyvä mekanismi — harvinainen tauti. Hyvin kehittynyt melko aikaisin, nopealla kurssilla ja usein johtaa täydelliseen sokeuteen nuorilla tai pikkulapsilla. Sen syy on geenimutaatioita CRB1 (kromosomi 1) ja SPATA7 (kromosomi 14), on myös harvinaisia muotoja sairauksien, jotka aiheutuvat virheellisestä muita geenejä. Patogeneesiin peittyvästi periytyvä muotoja retinitis pigmentosa ei ole tutkittu, on odotettavissa, osallistuminen proteiinien koodaavat yllä geenit alkionkehityksen aikana elinten vision.
Pigmentoitu retiniitti, jolla on X-sidottu perinnöllisyys — edustaa myös tätä geneettistä tautia. Useimmiten se johtuu puutteita geenien ja RP2 RPGR väistyvä luonne perinnöllinen lähetyksen. Tästä syystä tämän tyyppinen retinitis pigmentosa vaikuttaa vain pojilla, ei homologisia X-kromosomi. Nämä geenit koodaavat proteiineja, entsyymejä, aktiivisesti mukana metaboliaan verkkokalvon, joten niiden vika johtaa häiriöille kliinisesti ilmaistuna retinitis pigmentosa.
Pigmentin retiniitti, joka johtuu mitokondrioiden DNA: n mutaatioista, on harvinainen tämän taudin muunnos. Se periytyy vain äidin linjalta ja siirretään äidiltä jälkeläisille. Lääkärit-genetiikka ei ole vielä kyennyt tunnistamaan mitokondrio-DNA: n alueita, jotka mutatoivat tässä patologian muodossa.
On olemassa myös muita tämän tyyppisiä luokituksia — kliinisen kurssin, samanaikaisten epämuodostumien, patologian puhkeamisen iän (synnynnäinen, nuoruusiän) ja lukuisten muiden kriteerien mukaan. Tällä ehdolla ei ole tällä hetkellä yleisesti hyväksyttyä luokittelua, mutta kaikkien sairauksien muodon jakautuminen perintönsä mekanismin avulla pidetään parhaiten ja ymmärrettävänä, joka kattaa suurimman osan pigmentti-retiniitin kliinisistä ja geneettisistä lajikkeista.
Retinitis pigmentosan oireet
Kehittäminen retinitis pigmentosa voi alkaa missä iässä tahansa — ja väistyvä sukupuoleen sidottu muodossa tauti ilmaantuvat yleensä varhaislapsuudessa, kun taas jotkut autosomaalinen dominantti laji voi ilmetä aikuisiässä ja jopa vanhuuteen. Tyypillisesti yksi ensimmäisistä oireista on pimeän sopeutumisen ja verenvuotojen väheneminen, joka voi säilyä patologian ainoana ilmentymänä useita viikkoja (nopeasti edistyksellisiä muotoja) tai vuosia. Pigmenttivalmisteen jatkokurssilla sokeus kehittyy yöllä (niktalopia) normaalin päivän näkökyvyn tasolla. Näiden ilmentymien syy on valon havaitsemisesta vastuussa olevien sauvojen hallitseva rappeutuminen alhaisessa valaistuksessa.
Tulevaisuudessa retinitis pigmentosa on ominaista näkökentän kaventuminen ja syrjäisten alueiden (perifeerinen skotoma) menetykset. Tämä on myös jatkossa tangot, jotka sijaitsevat pääasiassa pitkin verkkokalvon reunoja. Vaikeissa tapauksissa tunnelin visio kehittyy, sen vakavuus putoaa merkittävästi, potilaat, joilla on pigmentti-retiniitti, tulevat vammautumaan. Degeneratiiviset muutokset vaikuttavat silmien aluksiin, mikä johtaa kartioiden tuhoutumiseen, linssien ja lasimaisen ruumiin läpikuultamattomuuteen, skleroiden ohenemiseen. Näiden prosessien kokonaisuus johtaa potilaan täydelliseen sokeuteen. Ei kuitenkaan jokainen muoto retinitis pigmentosa on tyypillistä tämän tuloksen — monet autosomaalinen hallitseva erilaisia taudin pitkään, hämäräsokeus ja ilmenevät vain ovat jonkin verran kaventuneet näkökentässä.
Diagnoosi retinitis pigmentosa
Havaitsemaan retinitis pigmentosa avulla silmänpohjan tutkimus, sähköinen, Elektro-Okulografia ja muut oftalmiset tutkimus, tutkimus suvussa potilaan, molekyyligenetiikan analyysit. Kun potilaan valituksia heikentää näköä illalla, sinun on tehtävä täydellinen oftalmologinen tutkimus. Pohjan päällä voi olla eristyneitä pisteitä (luupetkiä), jotka sijaitsevat verkkokalvon kehällä — ne ovat rasvaisen pigmentin kerrostumia. Koska progressiivinen pigmentti retiniitti tulee yhä enemmän, ne alkavat muodostaa lähempänä keltaista kohtaa. Kun ilmaistu psittakoosin taudin silmänpohjan määritettynä kaventuminen arteriolien, hiussuonien surkastumista, ja tulevaisuudessa — vahamainen surkastuminen näköhermon.
Viskositeetin leveyden mittaus pigmentti-retiniitissä paljastaa niiden samankeskisen kapasiteetin erilaisten vakavuusasteiden mukaan (riippuen taudin vaiheesta). Tämän patologian ominainen ilmentymä on myös sinisen värin herkkyyden väheneminen tritanopiaan asti, joka määritetään Rubkinin taulukoiden avulla. Elektroretinografian kuva pigmentti-retiniitin kanssa riippuu patologian vaiheesta — kaikkien aaltojen vähentämisestä ja päättyessä rekisteröimättömällä ERG: llä täydellisellä sokeudella. Elektro-oculographian tavoitteena on laskea Ardenin kerroin, joka on yleensä vähintään 180%. Pigmentti-retiniitin kanssa sen arvoa voidaan pienentää 100 prosenttiin ja jopa pienemmäksi.
Molekyyliset geneettiset tutkimukset ovat tarpeen lopullisen pigmentti-retinitis-diagnoosin vahvistamiseksi, lisäksi nämä tiedot voivat olla hyödyllisiä sairauden ennusteiden määrittämisessä. Tällä hetkellä laboratoriossa on saatavilla geneettisen diagnoosin menetelmiä yleisten patologisten muotojen osalta, jotka johtuvat geenien RP1, RP2, RPO, CRB1, SPATA7, RPGR ja muut mutaatiot. Nämä tutkimukset kattavat noin 70-80% kaikista retinitis pigmentosa -tapauksista, mutta monissa harvinaisimmissa muodoissa ei ole kehitetty geneettisiä diagnoosimenetelmiä. Tyypillisesti tässä tapauksessa diagnostinen tekniikka pelkistetään edellä olevien geenien sekvenssin suora- tai automaattiseen sekvensointiin.
Pigmentin retiniitin hoito ja ennuste
Tällä hetkellä kehitystyössä ja kliinisissä kokeissa kehitetään erityisiä menetelmiä retinitis pigmentosa -hoidon hoitamiseksi. On olemassa lupaavia tuloksia geeniterapian, kantasolujen ja muiden lääketieteellisten tekniikoiden käytöstä. Kliinisissä käytännöissä käytetään vain tukihoitoa, jonka tarkoituksena on hidastaa pigmentti-retiniitin ilmaisujen etenemistä. Tätä tarkoitusta varten käytetään A-vitamiinivalmisteita, jotka parantavat verkkokalvon trofiaa ja muita näkökentän rakenteita. Joissakin maissa verkkokalvotehosteita on kehitetty, niiden implantaatio vaikuttaa positiivisesti retinitis pigmentosa-potilaan visuaaliseen toimintaan. Kuitenkin monissa tapauksissa, erityisesti autosomaalisten resessiivisten ja sukupuolisidonnaisten tautien muodoissa, huolimatta kaikista terapeuttisista toimenpiteistä, peruuttamaton sokeus kehittyy.
Retinitis pigmentosan ennuste katsotaan yleensä epäsuotuisaksi, koska tauti etenee tasaisesti ja johtaa lopulta täydelliseen sokeuteen. Tämän tilan eri muodoissa vain oireiden esiintymisnopeus on erilainen: se on korkeampi autosomaalisissa recessive-lajikkeissa ja on merkittävästi alhaisempi dominanttien patologisten tyyppien kanssa. Tukihoito voi viivästyttää sokeuden alkamista keskimäärin 5-10 vuotta, mutta kliinisessä käytännössä ei ole muita terapeuttisia toimenpiteitä pigmentti-retiniitin osalta. Ehkäisy on mahdollista lääketieteellisenä geneettisenä neuvonantajana riskeissä oleville vanhemmille (potilaille, joilla on retinitis pigmentosa tai sen läsnäolo läheisissä sukulaisissa). On myös suositeltavaa käyttää aurinkolasit, jotka joissakin lähteissä,