Pfeifferin oireyhtymä
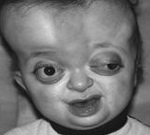
Pfayfferin oireyhtymä on geneettinen sairaus, jolla on autosomaalinen hallitseva perintämekanismi, jolle on tunnusomaista kallon ja raajojen luiden muodostumisen loukkaus. Oireet ovat kallon epämuodostumat (kraniosynostaosin seurauksena), sormien ja varpaiden luut. Joitakin taudin muotoja ovat luonteeltaan kuurous, henkisen kehityksen rikkominen, hammaslääketieteelliset sairaudet. Pfeifferin oireyhtymän diagnoosi tehdään potilaan nykyisen tilan, röntgentutkimusten ja molekyyligeneettisten analyysien perusteella. Ei ole erityistä hoitoa, palliatiivisia toimenpiteitä käytetään vakavien komplikaatioiden (mukaan lukien kirurgiset) ja oireenmukaisen hoidon estämiseen.
- Pfeifferin oireyhtymän syyt
- Pfeifferin oireyhtymän luokitus ja oireet
- Pfeifferin oireyhtymän diagnoosi ja hoito
Pfeifferin oireyhtymä
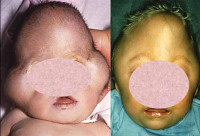
Pfayfferin oireyhtymä on synnynnäinen, geneettisesti määritelty kallon ja raajojen muodostumisen loukkaus, jolle on ominaista kraniosynostosis, kasvojen muodonmuutos, sormien laajentuminen ja lyhentäminen, usein syndaktyly. Saksan pediatri Rudolf Pfeiffer kuvasi ensimmäistä kertaa tämän taudin vuonna 1964, ja tulevaisuudessa patologian kliiniset muodot paljastuivat. Modernin genetiikan mukaan Pfeifferin oireyhtymä on autosomaalinen hallitseva tila, jossa täysi penetraatio, potilaiden sukupuolijakaumalla ei ole erityispiirteitä — sekä pojat että tytöt kärsivät samalla taajuudella. Tapahtumaa ei ole määritelty tarkasti joidenkin tietojen mukaan se on noin 1: 100 000 vastasyntyneelle. Tämän taudin jakautumisessa ei ollut kansallisia eikä rodullisia piirteitä.
Pfeifferin oireyhtymän syyt
Huolimatta siitä, että Pfeiffer oireyhtymä on perinnöllinen sairaus, jossa on autosomaalinen dominantti lähetyksen mekanismi, useimmissa tapauksissa tämän taudin ovat seurausta spontaani ituradan mutaatioita sukusoluissa vanhemmat. Tässä tapauksessa kahden geenin vika vastaa valtion kehitystä, niiden koodaamilla proteiineilla on samankaltaiset toiminnot. Ensimmäinen geeni, FGFR1, sijaitsee 8. kromosomissa ja koodaa reseptoriproteiinisekvenssin fibroblastin kasvutekijä-1: lle. Yleisin geneettisen virheen tyyppi tässä tapauksessa on missense-mutaatio 252Pro-Arg eksonissa 7a. Toisen geenin mutaatioita, jotka johtavat kehitystä Pfeiffer oireyhtymä, FGFR2, sijaitsee kromosomissa 10 — ilmentymistuote sen on myös reseptori fibroblastikasvutekijä (RFRF-2). Tässä geenissä on tunnistettu yli 26 erilaista mutaatiomuunnosta,
Johtuen siitä, että edellä mainitut proteiinit ovat hyvin samankaltaisia piirteitä, synnyssä oireyhtymä Pfeiffer on puutteita FGFR1 ja FGFR2-geeni on myös hyvin samanlainen. Seurauksena missensemutaatioita muuttaa ekspressoidun proteiinin rakenteen johdosta, jossa tuloksena reseptorin ei kykene täysin sitoutua aineen stimulaattori (fibroblastikasvutekijä) ja lähettämään tietoja soluun. Koska fibroblastit ovat aktiivisesti mukana prosesseissa luun muodostumista sikiön aikana ja ensimmäisen vuoden lapsen elämän, tällainen muutos reseptoreihin johtaa eri luun sairauksien.
Todiste tästä on se, että geenin puutteellinen FGFR1, lisäksi Pfeiffer oireyhtymä, johtaa trigonocephaly tyypin 1, ja useita muita häiriöitä ja FGFR2 — epäspesifinen craniosynostosis, crouzonin oireyhtymä ja muut vastaavat sairauksien. Kun Pfeiffer oireyhtymä, aiheuttaa ennenaikaista fuusio kallon luut, joka aiheuttaa muodonmuutoksen pää ja kasvot, kohonnutta kallonsisäistä painetta — tämä puolestaan voi aiheuttaa useita toissijaisia rikkomuksia neurosensoriset häiriöistä vakava henkinen jälkeenjääneisyys.
Pfeifferin oireyhtymän luokitus ja oireet
Tähän mennessä on jaettava vähintään kolme kliinisistä muodoista Pfeiffer oireyhtymä, jotka eroavat vakavuus rikkomuksia, suunta ja taudin ennusteen. Syy tähän ero on siinä, molekyyli- ja geneettisiä mekanismeja patologian — tyypin 1 aiheuttama yksittäinen mutaatio FGFR1 geenin (252Pro-Arg), ja jotkut FGFR2 rakenne häiriöitä, kun taas 2. ja 3. tyypin aiheuttamia eri suoritusmuotojen vikoja vain FGFR2-geenin. Parhaillaan tutkitaan taudin jokaisen muodon patogeneesin hienovaraisempia ominaisuuksia. Koska kliininen kuva on tyypillinen, asiantuntijat voivat määrittää erilaisia Pfeiffer-oireyhtymiä, jotka perustuvat vain patologian oireisiin. Molekyylinen geneettinen analyysi monissa tapauksissa on tarpeen vain diagnoosin lopulliseen vahvistamiseen.
Pfayffer-tyypin 1 oireyhtymä on taudin yleisin ja suotuisa prognostinen tapaus. Tällöin kasvojen poikkeavuudet havaitaan lapsen syntymästä — hypertelorismista, yläleuan kehittymisestä, laajentuneesta nenästä ja nenän takapuolesta. Joissakin tapauksissa voidaan havaita kovaa makua ja eksophthalmoja. Tällä Pfeifferin oireyhtymän muodolla kallon muotoiset muutokset pienenevät sen korkeuden ja pituuden lisääntymiseen, ja epäsymmetriaa on harvoin havaittu. Tulevaisuudessa hampaat voivat olla epänormaaleja — hampaiden kaarevuus, hypoplasia, taipumus kariisiin. Raajojen luiden epämuodostumat vähenevät käsien ja jalkojen ensimmäisten sormien halkeamien laajentamisella, osittainen syndykytys voi olla huomattava.
Pfayffer-tyypin 2 oireyhtymä on taudin vakavampi muoto, joka ilmenee huomattavassa määrin kallonsolujen fuusion kesken ja lukuisia muita poikkeamia. Pää, koska kraniosynostosis hankkii tunnusomaisen muoto «kottarainen» tai «apila lehti», on merkittävä hypoplasia keskellä kolmasosa kasvoista. Kallon muodostumisen törkeästä rikkomisesta johtuen tämäntyyppistä Pfeifferin oireyhtymää leimaavat voimakas henkinen hidastuminen ja useat neurologiset häiriöt. Liman kehityksen virheet vähenevät ensimmäisen sormen laajenemisen, syndaktyylisesti, kyynärliitosten ankyloosiksi. Lisäksi Pfayfferin tyypin 2 oireyhtymään liittyy usein sisäisten elinten erilaiset epämuodostumat. Kaikki edellä mainitut rikkomukset voivat johtaa kuolemaan varhaisessa iässä.
Pfayffer-tyypin 3 oireyhtymä on monessa suhteessa samanlainen kuin sen taudin edellisellä muunnoksella, mutta kraniosynostosi ei johda kallon muodonmuutokseen «apilanlehden» muodossa. Potilailla on pään korkeus, raajojen kehittymisvirheet (syndaktyly, nivelten ankylosis), eksophthalmos, neurologiset häiriöt. Pfeifferin oireyhtymän muunnokselle on tunnusomaista hampaiden varhainen kehitys, usein heidän kanssaan syntyneet vauvat (synnytyshampaat). Myös sisäisten elinten vikoja ja varhainen kuolema ovat mahdollisia sairauksien yhdistelmän vuoksi.
Tällainen erilainen kliinisen kuvan ja ennuste eri vaihtoehtojen Pfeiffer oireyhtymä vaikuttaa perintö taudin. Koska tyypin 1, erityisesti kun on kyse havaita ajoissa ja asianmukaista hoitoa, tunnettu siitä, että pysyvyys normaalin älykkyyden, hedelmällisyyden ja selviytymistä, voi olla autosomaalinen dominantti taudin leviämisestä jälkeläisiä. Raskaampia 2. ja 3. tyypit johtavat usein ennenaikaiseen kuolemaan tai syvällinen työkyvyttömyyden potilaiden, näistä vaihtoehdoista Pfeiffer oireyhtymän oireita vain seurauksena spontaani mutaatioita.
Pfeifferin oireyhtymän diagnoosi ja hoito
Pfayfferin oireyhtymän oikea-aikainen diagnoosi on äärimmäisen tärkeä, koska sen avulla voit tehdä ajankohtaisen palliatiivisen hoidon suunnitelman, välttäen komplikaatioita ja parantavan huomattavasti potilaan elämän laatua. Tämä pätee kuitenkin vain ensimmäisen tyypin taudille, koska patologian muissa variantteissa kehityshäiriöiden monimutkaisuus on niin vakava, että se ei käytännössä jätä mahdollisuuksia älyn säilyttämiseen ja tulevaisuudessa — potilaiden elämään. Pfeifferin oireyhtymän havaitsemiseksi käytetään ultraäänitekniikoita (mukaan lukien prenataalinen diagnoosi), radiografia, molekyyligenetiikka-analyysit. Prekohtaa ultraäänellä raskauden aikana tämän taudin läsnä ollessa lapsella 2-3 kuukautena, on mahdollista tunnistaa kallon muodostumisen loukkaus, syndykytys, sisäelinten epämuodostumat.
X-ray menetelmiä käytetään harvoin varhaisessa diagnosoinnissa Pfeiffer syndroomasta — tuottavat pääasiassa tutkimus kallo, joka havaitsee eriasteisia vakavuudesta craniosynostosis, hypoplasia leukaluun, että muoto muuttuu kiertoratojen. Myös röntgenkuvassa voit määrittää käsien ja jalkojen peukalon luiden muodon muutoksen. Molekyyligeneettinen diagnoosi on kliininen geneetikko — tässä tapauksessa, jos epäilevät olevansa Pfeiffer oireyhtymän tyypin 1 etsitään pistemutaatio 252Pro-Arg suoralla sekvensoinnilla eksonin 7 FGFR1 geeniä. Muissa tapauksissa, suorittaa sekvensointi 7. ja 9. eksonin FGFR2 geeni — joka on, jossa on suurin osa vikoja, jotka johtavat kehitystä Pfeiffer oireyhtymä. Avustavan roolin taudin diagnosoinnissa voi olla lääketieteellisen kuvantamisen menetelmiä,
Hoito Pfeiffer oireyhtymä on vain oireenmukaista ja palliatiivinen — asiantuntijat yrittävät eliminoida kohonnutta kallonsisäistä painetta, jotta normaali kehitys hermoston, lievittää häiriöt sisäelimissä. Tätä varten käytetään yleisesti kirurgisia tekniikoita — sormen erottelu, kallo muovi ja monet muut. Myös määrättyjä lääkkeitä, joilla pyritään parantamaan hermokudoksen tarjontaa — nootrooppisia huumeita, vitamiineja. Pfayffer-tyypin 1 oireyhtymän oikea-aikaisella hoidolla potilaiden selviytyminen kasvaa merkittävästi. Valitettavasti lääketieteellisen hoidon määrä ei useinkaan riitä potilaille, joilla on toisen ja kolmannen tyyppisiä Pfayfe-oireyhtymiä.
Pfeifferin oireyhtymän ennuste ja ehkäisy
Pfeifferin oireyhtymän ennustus määräytyy tämän taudin variantin mukaan. Ensimmäiselle tyypille on ominaista suhteellisen suotuisa ennuste, joka riippuu patologian ajallisuudesta ja valitun hoidon oikeellisuudesta. Suotuisissa olosuhteissa potilaat säilyttävät normaalin älykkyyden, elävät vanhuuteen ja voivat saada jälkeläisiä — samalla kun riski sairauden välittämisestä lapsille on vähintään 50 prosenttia. Toisen ja kolmannen tyypin Pfayfferin oireyhtymä kuitenkin on erittäin epäsuotuisa ennuste — neurologisten ja muiden sairauksien yhdistelmä aiheuttaa usein kuoleman varhaislapsuudessa. Vaikka potilailla on lievittäviä toimenpiteitä, havaitaan vaikeaa oligofreniaa ja syvää invaliditeettia. Pfeifferin oireyhtymän ehkäisy, kun otetaan huomioon tämän taudin kehittymisen usein spontaani mekanismi,