Perinnöllinen amyloidoosi
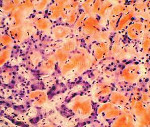
Perinnöllinen amyloidoosi — ryhmä sairauksia, joille on tunnusomaista kertyminen liukenemattoman proteiinin komplekseja (amyloidi) eri kudoksissa — pääasiassa hermoston ja ruoansulatusjärjestelmä, munuaisten ja sydänlihaksen. Oireet ovat neurologiset häiriöt, sydänsairaudet (kardiomyopatia), nefropatia ja dyspepsia. Erilaisten perinnöllisten amyloidoosien muodoissa vallitsee tietyn järjestelmän tappio. Diagnoosi perustuu kliinisiin ja histologinen tutkimus kudokseen joitakin muotoja menetelmien kehittämisen molekyyligenetiikan määritys. Hoitoon sisältyy immunosuppressiivisten lääkkeiden käyttö, tukeva ja oireinen hoito.
- Perinnöllisen amyloidoosin syyt
- Perinnöllisen amyloidoosin luokitus ja oireet
- Perinnöllisen amyloidoosin diagnosointi ja hoito
Perinnöllinen amyloidoosi
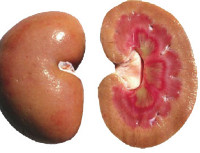
Perinnöllinen tai familiaalinen amyloidoosi — geneettisesti määräytyvä vastoin proteiinimetaboliaa joka on muodostelma epätavallista proteiinin ja sen myöhempi immuunikompleksien kerrostumisen koostumuksessa eri kudoksissa. Kuten nosological yksikkö amyloidoosi on tiedetty jo pitkään, mutta geneettinen luonne joissakin muodoissa ehto löydettiin vasta viime vuosikymmenellä. Erilaistumista perinnöllinen ja periytyvä muodot huomattavasti vaikeuttaa se, että useat geneettisesti määräytyvä patologioiden (esim., Määräajoin tauti) on myös tunnusomaista amyloidin kertymistä, mutta nyt oletetaan, että proteiini sellaisissa sairauksissa häiriöt ovat toissijaisia.
Tällä hetkellä ensisijainen perinnöllinen amyloidoosi luotettavasti sisältää ainoastaan sellaisia sairauksia johtuu mutaatioista TTR geenissä. Kiistanalainen kysymys on luokittelu ns amyloidoosi Suomen tyyppiä — totesi, että tämä tauti on aiheuttanut vika GSN geeni, mutta se on vielä epäselvää, amyloidikertymillä joiden ensisijainen häiriö tai seurauksena muita prosesseja. Joissakin geneetikot, lääkärit yhdistää tämän taudin tyypistä, joilla on perinnöllinen amyloidoosi tyyppi 4, mutta joilla on erilainen geneettinen luonteeltaan. Perinnöllisen amyloidoosin esiintymistä ei ole määritetty tämän tilan harvinaisuuden vuoksi.
Perinnöllisen amyloidoosin syyt
Välitön syy perinnöllinen amyloidoosi on ensisijainen puutteita TTR-geeni, joka sijaitsee 18. kromosomi. Tällä hetkellä tunnistetaan enemmän kuin 80 eri mutaatioiden tämän geenin, joista monet johtavat eri patologioiden — lisäksi kaikki muodot perinnöllinen amyloidoosi, nämä ovat distranstiretinemicheskuyu hyperthyroxinemia ja familiaalinen muodot rannekanavaoireyhtymä. Lähes kaikki TTR viat periytyvät autosomaalinen dominantti mekanismi, geenin ekspressio on proteiini transtyretiini suorittavat liikenteen proteiinin toimintoja tyroksiinin ja retinolia. On muitakin sairauksia, jotka johtuvat vahingot eri rakenteisiin ja muodostumista transtyretiiniä — mukaan lukien systeeminen seniili amyloidoosi ja amyloidipolyneuropatiaa.
Perinnöllisen amyloidoosin patogeneesi liittyy transtyretiinin molekyylien vakauden rikkomiseen — TTR-geenin mutaation vuoksi, tämän proteiinimolekyylin rakenne muuttuu ja se muuttuu epästabiiliseksi. Useissa tapauksissa epävakaus vaikuttaa vähäiseen toimintaan, mutta joskus molekyylin konformaatio muuttuu niin paljon, että se saa immunogeenisiä ominaisuuksia. Epänormaali proteiini aiheuttaa reaktiota, joka muistuttaa allergista (immunokompleksityyppiä), jolloin muodostuu liukenemattomia proteiinikomplekseja, jotka ovat alttiita kerrostumalle kudosten solunulkoisessa tilassa. Perinnöllisen amyloidoosin kliiniset ilmentymät riippuvat siitä, missä proteiinikompleksien hallitseva laskeuma aiheuttaa elinten toimivuuden rikkomisen.
Perinnöllisen amyloidoosin luokitus ja oireet
Perinnöllinen amyloidoosi johtuu mutaatioista TTR-geenin, joka kykenee ilmenee häiriöitä, eri elinten ja kudosten — hermoja, vatsa ja 12. tiperstnoj munuainen, sydän, silmä. Tällöin psittakoosin taudin riippuu mutaation luonne, eri muodoissa eroavat vallitsevaa tappion tahansa elinten vakavuudesta rikkomuksia ja koko nykyisen. Tähän mennessä on olemassa 6 suurta fenotyyppistä perinnöllistä amyloidoosia.
Tyyppi 1 (Andrade-oireyhtymä)— on perinnöllisen amyloidoosin yleisin variantti, kuvattiin ensimmäisen kerran vuonna 1952. Yleisimpiä Portugalissa, samoin kuin entisissä portugalilaisissa siirtomaissa (Latinalaisessa Amerikassa). Alkuperä tämäntyyppisen perinnöllinen amyloidoosi useimmiten kirjattu ikäryhmä 30-40 vuotta, ensimmäinen oire on vastoin herkkyys (kipu, lämpötila, tuntoon) iholla alaraajojen. Polyneuropatia on vähitellen voimistuvan, lisäksi tuhoutuminen tuntohermojen lopulta liittyä moottorin lihassairaus distaalisten jaloissa. Perinnöllisen amyloidoosin aiheuttama innervaation loukkaus johtaa usein alhaisten ääripäiden troofisiin haavaumiin. Patologia vaikuttaa myös autonomiseen hermostoon — potilaissa esiintyy voimakkuutta ja ortostaattista hypotensiota.
Tyypin 2 (Rukavin-oireyhtymä) on hyvin harvinainen perinnöllinen amyloidoosi, joka kuvattiin luotettavasti vain USA: n vuonna 1956 kahden perheen edustajista. Se ilmaantuu 35-45 vuoteen ja alkaa tunnetulla tunnelisoliongella, jossa muutaman vuoden kuluttua liittyy herkkien ja autonomisten hermojen osallistuminen. Tällainen perinnöllinen amyloidoosi etenee hyvin hitaasti, 15-20 vuoden kuluttua taudin puhkeamisesta, amyloidin talletukset suolessa, sydämessä ja munuaisissa.
Tyyppi 3 (Van Allen -oireyhtymä) on eräänlainen perinnöllinen amyloidoosi (perinnöllinen amyloidinen neuropatia), jolle on tyypillistä hermojen ja suolten yleinen vaurio. Tämän taudin kliiniset oireet ovat hyvin samankaltaisia kuin tyypin 1, mutta pääasiallinen ero on siinä tapauksessa, että autonomisessa hermojärjestelmässä ei ole merkittävää vahinkoa. Myös potilaat, joilla on tällainen perinnöllinen amyloidoosi, ovat usein 12-tyypin suoliston gastriitti ja haavaumat.
Suomalaista tyyppiä 4 tai perinnöllistä amyloidoosia kuvataan ensimmäisen kerran vuonna 1971, ja sen geneettinen luonne on jo käynnissä, kuten edellä on jo mainittu. Pohjimmiltaan tämä taudin muoto vaikuttaa kraniaalisiin hermoihin, yläraajojen herkkyys heikkenee. Ajan myötä on sarveiskalvon ristion atrofia ja ihon turgor vähenee, patologian kehityksen loppuvaiheissa on voimakas polineuropatia ja amyloidin kertyminen sydämessä. Toisin kuin muilla perinnöllisillä amyloidoosilla, tällainen tyyppi voi periytyä autosomaalisella resessiivisellä mekanismilla (jos tauti aiheutuu GSN-geenin mutaatioista).
Perinnöllinen kardiomiopatichesky amyloidoosi tai familiaalinen amyloidoosi tanskalainen tyyppi ilmenee lieviä neurologisia häiriöitä ja huomattavia sydänlihasvaurioon (amyloidoosi sydämen). Amyloidiesiintymät johtavat sydänlihaksen ja sydämen vajaatoiminnan nopeaan kehittymiseen, mikä aiheuttaa kuoleman.
Nefropatichesky amyloidoosi, kuurous, nokkosihottuma ja kuume — harvinainen, mutta melko vaikea perinnöllinen amyloidoosi, jossa potilasta iältään 20-30 vuotta, jonka tuntemattomasta syystä aiheutuva kuume, ja usein esiintyy nokkosihottuma. Sitten näihin ilmentymiin lisätään neurosensorinen kuulohäviö, jota myöhemmin vaikeuttaa täydellinen kuurous. munuaissairaus (munuaisten amyloidoosi) johtaa usein krooniseen munuaisten vajaatoimintaan, joka aiheuttaa kuoleman eniten uremiassa.
Perinnöllisen amyloidoosin diagnosointi ja hoito
Perinnöllisen amyloidoosin diagnoosi tehdään potilastutkimustietojen perusteella, yleiset kliiniset analyysit, tärkeimpien elinten (hermoston, sydämen, munuaisten) tilan arviointi, kudosten histologinen tutkimus, molekyylibiologiset tutkimukset. Tutkinnassa voidaan havaita neurologisten vaurioiden merkit (aistien ja motoristen toimintojen häiriöt, hypo- tai areflexia), pitkittyneissä tapauksissa lihasten atrofia on havaittavissa innervaation puutteen vuoksi. Silmätaudit, joilla on perinnöllinen amyloidoosi, paljastavat usein amyloidikertymän merkkejä vitreäävässä huumorissa, sarveiskalvon atrofia tai haavauma on mahdollista. Joissakin tapauksissa voi olla ristiriitoja iholta — turgorin, urtikarian, troofisten haavaumien väheneminen.
Sydän- ja munuaisten ultraäänitutkimus, jolla on perinnöllinen amyloidoosi, antaa usein paljon tietoa tästä taudista. Amyloidin kertymisen munuaisissa, ne lisäävät ja kompaktoivat rakenteen avulla, joskus määrätään kystien läsnäolo. Amyloidoosin lopullisissa kehitysvaiheissa munuaiset voivat vähentyä niiden atrofian vuoksi. EKG: ssä kaikkien hampaiden amplitudi ja jännite pienenevät, ekokardiografia osoittaa sydänlihaksen paksuuden ja vasemman kammion koon merkittävää kasvua. Useissa tapauksissa ultraäänitutkimus, jolla on perinnöllinen amyloidoosi, voi paljastaa amyloidin talletukset muissa elimissä — maksa, perna, suuret astiat. Kun perinnöllisestä amyloidoosista kärsivien potilaiden potilaiden biopsia havaitaan tyypillinen histologinen malli — eosinofiilisten massojen kertyminen ja konsentraatio verisuonien ympärillä.
Perinnöllisen amyloidoosin erityistä hoitoa ei ole, pääasiassa oireenmukaista ja tukevaa hoitoa käytetään. Taudin kulun hidastumiseksi käytetään immunosuppressiivisia aineita (erityisesti kortikosteroideja) sytotoksisten lääkkeiden määräämiseen. Jos perinnöllistä amyloidoosia aiheuttavat merkittävät munuaisten vauriot, hemodialyysi on osoitettu. Oireellinen hoito on myös tarpeen kardiomyopatian ja sydämen vajaatoiminnan varalta. On suositeltavaa ottaa suurempia annoksia B-vitamiineja, sillä ne hidastavat kehitystä neuropatian ja auttaa parantamaan yleistä potilaan.
Perinnöllisen amyloidoosin prognoosi
Useimmissa tapauksissa ennuste on epäsuotuisa perinnöllinen amyloidoosi, kuten monet tautimuodoille johtaa kuolemaan 10-25 vuotta sen jälkeen, kun se alkoi. Oireenmukaista hoitoa, hemodialyysi, immunosuppressiiviseen aineet voivat hidastaa kehitystä tämän patologian. Koska useimmat oireet perinnöllisen amyloidoosin nähty 30-40 vuotta, diagnosoida ajoissa ja älykäs ylläpitohoito voi tarjota selviytymisen vanhuuteen ilman merkittävää heikkenemistä elämänlaatua.