Hunterin oireyhtymä
Hunterin oireyhtymä — perinnöllinen sairaus metabolinen X-kromosomiin liittyvä periytyy resessiivisesti, tunnettu siitä, lysosomaalisen entsyymin, iduronaatti-2-sulfataasi ja kertymistä mucopolysaccharides kudoksissa. Kun Hunterin oireyhtymä on merkitty kasvun hidastuminen, macrocephaly muodonmuutos osteoarticular laitteiston, ihovaurioita, sydän- ja hengityselimiin, hepatosplenomegalia, kuulon heikkenemistä, kehitysvammaisuus. Jotta voidaan diagnoosi Hunterin oireyhtymä kuullaan genetiikka, määritelmä glykosaminoglykaanien erityksen, radiografian luiden ja nivelten. Hunterin oireyhtymää sairastavilla potilailla on esitetty elinikäinen entsyymikorvaushoito elapratilla.
- Hunterin oireyhtymän syyt
- Hunterin oireyhtymän luokittelu
- Hunterin oireiden oireet
- Hunterin oireyhtymän diagnosointi
- Hunterin oireyhtymän hoito
- Hunterin oireyhtymän ennuste ja ehkäisy
Hunterin oireyhtymä

Hunter oireyhtymä (mukopolysakkaridoosin tyyppi II) — harvinainen geneettinen sairaus, ketjutetaan X-kromosomin, jossa epäonnistuminen tapahtuu, koska osittaisen entsymaattisen hajoamisen ja kertymistä happamien mukopolysakkaridien (glykosaminoglykaanit) eri kudoksissa. Hunterin oireyhtymän esiintyvyys on noin 1: 100 000-150000 vastasyntyneellä. Tällä hetkellä Hunterin oireyhtymässä on enempää kuin 2000 potilasta maailmassa; Venäjällä ei ole virallisia tilastoja. Hunterin oireyhtymä viittaa ryhmään orpojen sairauksia, joiden hoito, nykyisen lainsäädännön mukaan olisi rahoitetaan liittovaltion ja aluebudjettien.
Hunterin oireyhtymän syyt
Kehitys Hunterin oireyhtymään liittyy geenin mutaatio iduronatsulfatazy (IDS), joka koodaa lysosomaalisen entsyymin, on iduronaatti-2-sulfataasi. IDS-geeni on kartoitettu paikkakunnalla Xq28 X-kromosomin pitkän haaran kohdalla; Tällä hetkellä tunnetaan yli 150 erilaista mutaatiota (pistemutaatiot, pienet ja suuret deleetioita, insertioita, uudelleenjärjestelyjä jne.).
Koska X-kromosomin perinnöllisyys on ketjutettu, Hunterin oireyhtymä vaikuttaa yleensä vain poikia (XY). Heterotsygoottinen naiset ovat useimmissa tapauksissa tyypin II mukopolysakkaridoosin harjoittajat ilman kliinisiä oireita, mutta useissa tapauksissa on kuvattu Hunterin oireyhtymä tytöillä, jotka liittyvät uuden mutaation tai inaktivaatio toinen normaali X-kromosomi.
Mutaatiot IDS geeni liittyy puute tai entsyymin puute iduronaatti-2-sulfataasi (I2S), epätäydellinen pilkkominen ja kertymistä lysosomeihin solujen käytännöllisesti katsoen kaikkien kudosten ja elinten glykosaminoglykaanien (GAG: t) — dermataanisulfaatti ja heparaanisulfaatin.
Hunterin oireyhtymän luokittelu
Hunterin oireyhtymän kliinisten ilmiöiden vakavuus (kohtalainen tai vaikea) riippuu IDS-geenin mutaation tyypistä. Siten suuret deleetio- ja kompleksiset uudelleenjärjestelyt aiheuttavat pääsääntöisesti Hunterin oireyhtymän vaikeiden muotojen kehittymisen. Vakavien (Mukopolysakkaridoosi tyyppi II A) on ominaista varhainen kehitys kliinisten oireiden (lähinnä vuotiaista 18-36 kuukautta), nopeasti etenevä, vaikea kehitysvammaisuus, monielinhäiriö. Potilaat, joilla on vaikea Hunterin oireyhtymä, kuolevat elämässä toisella vuosikymmenellä.
Keskipitkä raskas muoto (mukopolysakkaridoosi tyyppi IIB) on noin 1/3 kaikista patologiatapauksista. Hunterin oireyhtymän kliiniset oireet ilmenevät tavallisesti lapsilla 3-8 (joskus 10-13) vuotta; äly on yleensä säilynyt; Elinajanodote suotuisissa olosuhteissa voi nousta 50-60 vuoteen. Hunterin oireyhtymän lievää muotoa sairastavat potilaat voivat menestyksekkäästi hyötyä ammattikentällä ja olla terveitä jälkeläisiä.
Hunterin oireiden oireet
Synnytyshetkellä Hunterin oireyhtymän lapset näyttävät kliinisesti terveenä. Tärkein oireyhtymä on keskimäärin 2-4 vuotta. Ennen tätä ikää tautitapaukset ovat epäspesifisiä: lapsilla voi olla toistuva nuha, meluisa hengitys, napanuorat ja nenäverenvuoto, hydrocele.
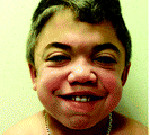
Aikaisin ominaispiirre Hunterin oireyhtymä on asteittainen muutos ulkonäön lapsi: kasvojen piirteet tulevat karkea (gargoilizm); suu, huulet ja sieraimet — suuri; iho on paksu. Look potilas Hunterin oireyhtymää täydennetty lyhytkasvuisuus, lisääntynyt koko pään (macrocephaly), lyhyt kaula, epänormaalit hampaat (hampaat harvinainen). Lapset Hunterin oireyhtymä on ulospäin hyvin lähellä toisiaan, ja muistuttaa veljekset.
Muihin tyypin II mukopolysakkaridoosiin liittyviin varhaisiin oireisiin kuuluvat röyhkä matala ääni, suurentunut vatsa, hepatosplenomegalia. Lapset, joilla on Hunterin oireyhtymä, ovat alttiita akuuttien hengitysteiden virusinfektioiden, otitis mediaa, kurkunpäänsärkyä, trakeittia, keuhkokuumetta; usein heillä on obstruktiivinen uniapnea, krooninen ripuli. Seurauksena glykosaminoglykaanien ja lipidien kerääntymisestä dermaalissa on nodulaaristen papulaaristen ihottumien muodostuminen hartioiden, lantion ja olkapäiden ihoon. Ehkä syntyy «Mongolian paikat» lumbosakraalisella alueella, hypertrikoosi.
Jo kouluikäisten lasten Hunterin oireyhtymää, on merkkejä tappion muskuloskeletaalisysteemin johtavat nivelten jäykkyys, kyphoscoliosis kehittäminen, vääntymistä harjat tyyppiä «kynnet.» Liikkeet muuttuvat vaikeiksi, käynti äkillisesti; nuoruudelle, Hunterin oireyhtymää sairastavat potilaat ovat usein vammattomat invalidit, bedridden.
Neurologiset häiriöt johtuvat Hunterin oireyhtymää sisältävät ylivilkkaus oireyhtymä, kouristukset, viestintään vesipää, spastinen paraplegia, kieli viive, progressiivinen kuulon heikkenemistä. Visuaalisen järjestelmän puolella havaitaan sarveiskalvon läpinäkyvyys ja epätyypillinen retinitis pigmentosa. Viimeisimmän merkkejä mukopolysakkaridoosin tyypin II kuuluvat Sydän: syntyminen sivuääniä, osti sydäntaudit (useimmiten hiippaläpän vajaatoiminta), kardiomyopatia, ja toiset.
Lapsi saattaa kokea muutoksia psyykeissä: herkkyys, aggressiivisuus. Hunterin oireyhtymän tyypistä riippuen älyllinen vika voi olla pieni tai merkittävä. Lapsellinen tulos potilailla, joilla on Hunterin oireyhtymä, tulee yleensä progressiivisesta sydämestä tai keuhkojen vajaatoiminnasta.
Hunterin oireyhtymän diagnosointi
Pediatrisessa käytännössä Hunterin oireyhtymä on erittäin harvinaista. Kuitenkin Mukopolysakkaridoosi tyypin II voidaan epäillä perusteella kliinisten oireiden (ulkoiset muutokset, taudin manifestaatioita 2-4 elinvuoden, ja muut edistykselliset tietenkin.). Diagnoosin vahvistamiseksi lasten on kuultava geneetikkoa, analysoitava kliinisiä genealogisia tietoja ja suoritettava molekyylibiologisia tutkimuksia.
Tärkeä biokemiallinen markkeri Hunterin oireyhtymä kasvaa eritystä mukopolysakkaridien (dermataanisulfaatti, heparaanisulfaatti) virtsassa, entsyymin puutos iduronaatti-2-sulfataasi. Röntgentutkimukset kallon luut, nivelet, pitkät luut, selkärangan osoittavat dysostoosi, nivelrikko, useita muutoksia nikamien.
Havaita morfologisia ja toiminnallisia muutoksia sisäelinten ja keskushermoston suorittaa vatsan ultraääni, elektrokardio- grafia, kaikukardiografia, electroencephalography, MRI. . Morfologinen tutkimus kudosten biopsialla saatu ihon, maksan, sydänlihas, jne., Havaitsee muutokset samaa tyyppiä — solujen täytetty glykolipidit.
Perheissä tunnetun genotyypin ja suuri riski saada lapsi Hunterin oireyhtymää voidaan suorittaa invasiivisen sikiödiagnostiikkaa — Korionvillusbiopsia, amniocentesis tai cordocentesis määritelmän aktiivisuuden iduronaatti-2-sulfataasi, että tuloksena oleva materiaali.
Erotusdiagnoosissa Hunterin oireyhtymä olisi suorittaa muita mukopolysakkaridoosin (erityisesti Hurler oireyhtymä), sekä muut lysosomaalisiin.
Hunterin oireyhtymän hoito
Hunterin oireyhtymällä varustetuilla lapsilla on oltava elinikäinen fermentin korvaushoito. Tähän mennessä ainoa huume, joka on rekisteröity Yhdysvalloissa, Euroopassa ja Venäjällä Hunterin oireyhtymän hoidossa, on idursulfaasi. Potilaat, joilla on tyypin II mukopolysakkaridoosin Idursulfaasi tulisi saada viikoittain tiputuslaitteessa annoksella 0,5 mg / kg kehon paino.
Sitä paitsi patogeneettisiin hoito, lasten Hunterin oireyhtymää, säännöllistä lääkitystä hinnat oireenmukaista ja korjaavien terapian Hepatonprotektorit, vitamiineja, antioksidantteja, cytoprotector. Monimutkainen hoito Hunterin oireyhtymä on aiheellista sisällyttää liikunta, fysioterapia (elektroforeesi lidazy nivelet, parafiinia kylpyjä, magneettiterapia, Laser punktio), istuntojen puheterapeutin ja defectology.
Hunterin oireyhtymän ennuste ja ehkäisy
B-tyypin Hunter-oireyhtymän suotuisampi kurssi ja ennuste; ajallaan ja säännöllisellä hoidolla potilaiden elinajanodote voi nousta 50-60 vuoteen. Tyypin II mukopolysakkaridoosin vakavissa muodoissa potilaat kuolevat yleensä ennen 20-vuotiaita sydän- ja verisuonitaudista. Tähän mennessä vakava ongelma Hunterin oireyhtymää sairastavilla potilailla on idursulfaasin elintärkeän valmisteen oikea-aikainen vastaanottaminen korkeiden kustannusten vuoksi.
Lapset Hunterin oireyhtymää tarpeen seurata useiden asiantuntijoiden: lasten genetiikka, lastenlääkäri, lasten kardiologi, neurologi lasten epileptologa, lasten silmätautien, lasten otolaryngologist, lastenkirurgia, lasten ortopedia.
Tärkein tapa estää Hunterin oireyhtymä on geneettisen neuvonnan pareille todennäköisyys sairaan lapsen syntymää, sekä suorittamalla ennen syntymää diagnoosi. Sinun pitäisi tietää, että Hunterin oireyhtymää sairastavilla syntyy terveitä poikia ja tyttäriä palvella obligantymi kantajia mutantti geeni.