Hemoglobinopatioille
Hemoglobinopatiat ovat ryhmä vakavia perinnöllisiä verisairauksia, jotka johtuvat hemoglobiinin rakenteen rikkomisesta tai yhden tai useamman globiiniketjun synteesin vähenemisestä. Kliininen kuva on erittäin monipuolinen. Yleisiä oireita ovat hemolyyttinen anemia, pernan laajentuminen, luun vaurioituminen. Diagnoosi suoritetaan käyttämällä perifeerisen veren tahran, hemoglobiinielektroforeesin, geneettisten tutkimusten mikroskopiaa. Hoitoon käytetään veren komponenttien, hydroksiureavalmisteiden ja infuusioterapian siirtoa. Vaikeilla potilailla suoritetaan splenektomia, kantasolujen allotransplantaatio.
- Hemoglobinopatioiden syyt
- synnyssä
- luokitus
- Hemoglobinopatian oireet
- komplikaatioita
- diagnostiikka
- Hemoglobinopatian hoito
- Ennuste ja ennaltaehkäisy
Yleistä tietoa
Hemoglobinopatiat ovat joukko synnynnäisiä hemolyyttisiä anemioita, joille on tunnusomaista hemoglobiinin aminohapposekvenssin muutos tai globiiniketjujen muodostumisen estäminen. Nämä patologiat päättyvät usein kuolemaan jo varhaislapsuudessa. Noin 50 hemoglobinopatian tyyppiä tunnetaan. Yleisimpiä ja hengenvaarallisia pidetään sirppisolun anemiaa (SKA) ja talassemiaa. Hemoglobinopatiat ovat yleisiä Keski-Afrikan alueella, Etelä-Aasiassa, ja niitä havaitaan pääasiassa negroidessa. Talassemiaa esiintyy myös Välimeren maissa. Noin 350000 lasta, joilla on hemoglobiinivika, syntyy vuosittain.
 hemoglobinopatioille
hemoglobinopatioilleHemoglobinopatioiden syyt
Hemoglobinopatiat ovat autosomaalisia resessiivisiä geneettisiä tauteja. Kvalitatiiviset hemoglobinopatiat kehittyvät geenien mutaatioiden seurauksena, jotka ovat vastuussa tiettyjen aminohappojen synteesistä globiinin beta-ketjussa. Tämän seurauksena yksi aminohappo korvataan toisella (glutamiinihappo valiinille, lysiinille jne.). Tämä johtaa epänormaalin hemoglobiinin muodostumiseen, joka on paljon vähemmän liukoinen kuin normaali hemoglobiini A, jolloin punaiset verisolut ovat eri muodossa (kohdemuotoinen, sirppimäinen), joka rikkoo niiden toimintoja ja vähentää elinajanodotetta.
Kvantitatiiviset hemoglobinopatiat johtuvat geenien mutaatiosta, jotka koodaavat koko globiinin ketjua (useimmiten alfa- ja beeta). Tässä tapauksessa globiiniketjujen välinen tasapaino muuttuu — alfa-ketjujen riittämättömällä synteesillä tapahtuu ylimäärä beeta-ketjuja ja päinvastoin. Erytrosyyttien koko pienenee, niiden hemoglobiinipitoisuus pienenee ja kalvo muuttuu herkemmäksi erilaisille vaurioille.
On olemassa tekijöitä, jotka aiheuttavat vakavia hyökkäyksiä (kriisejä). Näitä ovat dehydraatio, hypotermia, infektio, johon liittyy korkea kuume. Naisilla paheneminen ilmenee usein raskauden taustalla. Korkealaatuisen hemoglobinopatian pääasiallinen patologinen ärsyke on veren happipitoisuuden väheneminen (hypoksia). Tämä voi tapahtua esimerkiksi nousemalla korkeammalle korkeudelle (kiipeily vuorella, lentäen lentäen), jossa hapen osapaine paineessa ilmaan vähenee tai jos hengityselinten vakavia sairauksia (keuhkokuume) esiintyy.
synnyssä
Hemoglobinopatioilla on samanlaiset patogeneettiset mekanismit. Hemoglobiinin muuttunut rakenne ennakoi voimakasta hemolyysiä. Pitkäaikainen nykyinen anemia edistää luuytimen hyperplasiaa. Kallon luut ovat muodonmuutoksia, selkärangan kaarevuus. Hematopoieesin ekstramedullariset fokukset kehittyvät, mikä johtaa maksan ja pernan (hepatosplenomegalia) koon kasvuun. Splenomegaliasta johtuu hypersplenismi — punasolujen lisääntynyt tuhoaminen pernasinusoidien avulla.
Punasolujen säännöllisen hemolyysin vuoksi maksa erittää suuren määrän bilirubiinia sappeen, mikä luo edellytykset sappirakon kivien muodostumiselle. Potilaat, joilla on hemoglobinopatioita, kokevat usein raudan ylikuormitusta sekä jatkuvan verensiirron vuoksi että raudan imeytymisen lisääntyessä ruoansulatuskanavassa. Suuri määrä rautaa kudoksissa lisää lipidiperoksidaatiota, joka vahingoittaa erilaisia elimiä.
Laadukkailla hemoglobinopatioilla veren vähentyneen happipitoisuuden vaikutuksesta liukenemattoman epänormaalin hemoglobiinin molekyylit venyttävät erytrosyyttikalvoa, mikä johtaa niiden muodon muutokseen. Deformoidut erytrosyytit kuljettavat happea huonommin ja kykenevät myös tarttumaan verisuonten endoteeliin, jolloin tukkeutuvat pienet astiat, jotka aiheuttavat tromboosia, tukkeutumista ja sydänkohtausta.
luokitus
Hemoglobinopatiat on jaettu kvalitatiivisiksi hemoglobiinin rakenteen (aminohapposekvenssin) rikkomisen ja kvantitatiivisen vuoksi, jolle on tunnusomaista globiiniketjujen muodostumisen väheneminen. Kvalitatiiviset hemoglobinopatiat esitetään seuraavilla muodoilla:
- Sirppisolun anemia (hemoglobinopatia S) . Yleisin tyyppi. Se on jaettu homotsygoottiseen muotoon (itse asiassa SKA), jossa on kirkkaat kliiniset oireet ja heterotsygoottinen kuljetus (sirppisolun poikkeama), oireetonta tai lievää kurssia.
- Hemoglobinopatia C. Lääkäriasema on samanlainen kuin SKA, mutta se on vähemmän selvä. Sen splenomegalia on suurempi kuin SKA: lla.
- Hemoglobinopatia CS (afrikkalainen reuma) . Adrift muistuttaa SKA: ta. Paroxysmal luu ja nivelkipu vallitsevat.
- Hemoglobiini E ja D . Jatka hieman hemolyyttistä anemiaa.
- Perinnöllinen metemoglobinemia. Tässä lajissa muodostuu useiden tekijöiden vaikutuksesta hemoglobiinin, metemoglobiinin, hapettunut muoto, joka on tiukemmin sitoutunut happeen eikä anna periksi kudoksilleen.
- Epävakaan hemoglobiinikantajan aiheuttama anemia. Tämä on hyvänlaatuinen patologia, jossa sulfa-lääkkeiden ottamisen jälkeen on lievä hemolyyttinen anemia.
Hemoglobiinin kvantitatiiviset poikkeamat ovat:
- Beetatalassemia. Yleisin vaihtoehto. Se on jaettu pieniin talassemiaan (heterosygoottinen kuljetus, jossa on oireeton kurssi tai lievä hemolyyttinen anemia) ja suuri talassemia (Cooleyn anemia), jolla on vakava kliininen kuva.
- Alfa-talassemia. Kurssi voi olla erilainen riippuen mutanttigeenien määrästä. Enimmäkseen samanlainen kuin heterotsygoottinen beeta-talassemia.
- Sikiön turvotuksen oireyhtymä hemoglobiini Bartilla (Hb Bart’s). Vakavin alfa-talassemian tyyppi. Lapsi kuolee kohdussa.
- Hemoglobinopatia H . Alfa-talassemian edullinen oligosymptomaattinen muoto.
- Beta-delta-talassemia. Käytännöllisesti katsoen erottamaton beta-talassemiasta.
- Hemoglobinopatioille Lepore . Tämä muoto kehittyy globiinibetaketjujen fuusion seurauksena ja on samanlainen kuin beta-talassemia.
Hemoglobinopatian oireet
Hemoglobinopatioiden kliininen kuva vaihteli. Heterosygoottisilla potilailla on joko oireeton tai lievä. Homotsygoottiset muodot alkavat näkyä jo jo varhaislapsuudesta (6 kuukautta — 1 vuosi). Yleisiä oireita ovat merkit hemolyyttisestä anemiasta (ihon ja ihon keltaisuus ja limakalvot, pernan laajentuminen), luuston kehityspatologia — tornikallo (nelikulmainen), litistetty nenän silta, kaareva selkä.
Bilirubiinin lisääntyneen erittymisen vuoksi sappeen sappikalvon oireita voi häiritä jo lapsuudessa — raskaus tai kipu oikeassa hypochondriumissa, sappikolbin aallot, ulosteen värjäytyminen. Pitkäkestoinen haavauma esiintyy usein jalkojen iholla. Sairauksiin, joissa on hemoglobiinirakenteen vika, jolle on ominaista raikas virtaus. Vakavimmat hyökkäykset, jotka usein päättyvät kuolemaan, esiintyvät sirppisolun anemiassa.
Vagoseclosure-kriisi
Tyypillisin. Eri elinten pieniä aluksia suljetaan. Lapset kokevat kipua pitkissä tubulaarisissa luissa, kädet ja jalat turpoavat, jolloin ranne, nilkanivelet (käsi-jalka-oireyhtymä) liikkuvat vaikeaksi. Suolistojen mikrotromboosit aiheuttavat vatsakipua. Nuoret miehet kehittävät usein priapismia, joka johtaa myöhemmin erektiohäiriöön. Kriisiin liittyy kuume, takykardia, hikoilu.
Hemolyyttinen kriisi
Hemolyyttisessä kriisissä punaisten solujen tuhoaminen on massiivinen ja hemoglobiinin ja punasolujen pitoisuus laskee jyrkästi. Olemassa oleva keltaisuus, ihon paljaus, limakalvot lisääntyvät, kuume, lanne- ja vatsakipu, verenpaineen laskun oireet (huimaus, pyörtyminen) liittyvät. Virtsa saa tumman värin suuren hemoglobiinimäärän vuoksi.
Sequestration-kriisi
Säilytyskriisin aikana myös hemoglobiinipitoisuus laskee nopeasti. Mutta tämä ei johdu hemolyysistä, vaan laskimotromboosista ja suurten veren määrien kertymisestä maksan ja pernan laajennettuihin sinusoideihin, mikä johtaa yleisen verenkierron köyhtymiseen. Vakavan anemian oireiden lisäksi vakava hepatosplenomegalia aiheuttaa vakavaa ja tuskallista kipua vasemmassa ja oikeassa hypokondriumissa.
Aplastinen kriisi
Varsin harvinainen kriisin muoto. Se kehittyy infektoituna parvoviruksella B19, joka kykenee estämään luuytimen verenvuotoa. Tähän liittyy nopea (mutta palautuva) väheneminen paitsi punasolujen, myös kaikkien muiden perifeeristen verisolujen (leukosyyttien, verihiutaleiden) pitoisuudessa. Siksi hemorraaginen oireyhtymä (verenvuoto nenästä, ikenet), erilaiset infektiot (lähinnä ARVI) liittyvät anemian oireisiin.
komplikaatioita
Hemoglobinopatioiden yleisiä komplikaatioita ovat JCB, pitkiä putkimaisia luut patologisia murtumia. Heterosygoottisilla muodoilla on harvoin haittavaikutuksia, sillä niillä on lievä kurssi. Kun kvantitatiiviset hemoglobinopatiat johtuvat ylimääräisen raudan kerääntymisestä sisäelimissä, sydämen vajaatoiminta, kirroosi ja tyypin 2 diabetes kehittyvät.
Hemoglobiinin kvalitatiivisille patologioille on tunnusomaista laaja valikoima haittavaikutuksia. Vaarallisimpia ovat keuhkoembolia, sydäninfarkti, aivohalvaus, joka noin 10%: lla potilaista johtaa kuolemaan. Luut johtavien mikrovälien tukkeutuminen johtaa reisiluun pään (ANGBK) aseptiseen nekroosiin. Pernan jatkuvan infarktin takia tapahtuu funktionaalinen asbestismi, minkä vuoksi bakteeritartunnat kehittyvät usein (keuhkoputkentulehdus, keuhkokuume) vakavalla kurssilla, usein kuolemaan.
diagnostiikka
Hematologia ja geneettiset lääkärit harjoittavat hemoglobinopatioita sairastavien potilaiden kuratointia. Yleiskatsauksen aikana kiinnitetään huomiota ihon väriin (pallor, keltaisuus), perustuslaillisiin loukkauksiin (lapsen hermostunut, lapsen fyysinen kehitys, luurankorakenteen poikkeavuudet). Lisätesti sisältää:
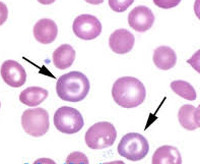
- Verikokeet. Tyypillinen kohtalaisen vakava normosyyttinen anemia on tyypillinen yleisiä verikokeita varten potilailla, joilla on kvalitatiivinen hemoglobinopatia, ja vakava mikrosyyttinen anemia on tyypillinen potilaille, joilla on kvantitatiivinen hemoglobinopatia. Verinäytteessä havaitaan muuttuneita deformoituneita erytrosyyttejä (sirppimäinen, pallomainen). Veren biokemiallisessa analyysissä vapaan raudan, ferriitin ja epäsuoran bilirubiinin pitoisuudet lisääntyvät.
- Hemoglobiinielektroforeesi. Kliinisessä hematologiassa laajalti käytetty hemoglobiinipatologioiden diagnosoinnin päämenetelmä, joka määrittää hemoglobiinifraktioiden suhdeluvun, on elektroforeesi kalvolla, jossa on selluloosa-asetaatti- tai sitraatti-agaria. Korkea epänormaalien hemoglobiinien (HbS, HbF, HBA2) pitoisuus havaitaan.
- Geneettiset tutkimukset. DNA-testaus PCR: llä suoritetaan vain osana synnytystä edeltävää diagnoosia, jonka tavoitteena on geneettinen neuvonta perheille, joilla on lisääntynyt riski sairastua hemoglobiiniin. DNA-näytteet saadaan amniosentesilla 8-10 ja 14-16 viikon raskausviikolla. Määritä eri geenien mutaatioiden läsnäolo.
- Instrumentaalitutkimukset. Raajojen luiden röntgenkuvauksessa havaitaan luuytimen lisääntymisen oireita — kortikaalisen kerroksen harventuminen, osteoporoosialueet, medullarikanavan laajentuminen. Kallon luut röntgensäteillä havaitaan neulamainen periostosis (”karvainen kranoli-ilmiö”). Vatsanontelon ultraäänitutkimus paljastaa hepatosplenomegalia, sappikiviä.
Hemoglobinopatiat erottelevat muiden synnynnäisten hemolyyttisten anemioiden kanssa (kalvopöydät, fermentaatiot, Minkowski-Chauffardin mikropallosairaus). Pysyvä tromboosi on erotettava eri trombofiliasta. Raudan ylikuormitus olisi erotettava perinnöllisestä hemokromatoosista. Anemia, ossalgia vaatii pahanlaatuisten myeloproliferatiivisten sairauksien poistamista.
Hemoglobinopatian hoito
Hemoglobinopatioita sairastavat henkilöt vaativat monimutkaisen monitahoisen hoidon, joten kaikki homotsygoottiset potilaat joutuvat sairaalahoitoon pakollisesti. Potilaita, joilla on heterotsygoottisia hoitomuotoja, ei ole ilmoitettu. Hemoglobiinin laadullisten ja kvantitatiivisten patologioiden ylläpitämisen perusperiaatteet eroavat jonkin verran toisistaan.
Konservatiivinen hoito
Konservatiivisen hoidon valinta tehdään ottaen huomioon hemoglobinopatian tyyppi, taudin kulku, tiettyjen komplikaatioiden esiintyminen. Arvioidaan sekä kliinisiä oireita että laboratoriotietoja (lähinnä punasolujen indeksejä). Hoitoalueita ovat seuraavat:
- Kriisien helpottaminen. Vasokoklusallisissa kriiseissä käytetään kipulääkkeitä (ei-steroidisia tulehduskipulääkkeitä) ja vakavan kivun oireyhtymän — huumausaineiden kipulääkkeitä. Myös deformoituneiden erytrosyyttien saostumisen estämiseksi ne määrätään hengittämällä hapella, oraalisesti tai laskimoon.
- Varoituskriisit. Hydroksiureaa määrätään potilaille korkealaatuisen hemoglobinopatian saamiseksi. Tämä lääke stimuloi sikiön hemoglobiinin muodostumista, joka tukahduttaa epänormaalien liukenemattomien hemoglobiinien synteesistä vastaavan geenin ilmentymisen, mikä vähentää punasolujen taipumusta muodonmuutokseen, vähentää kriisien esiintymistiheyttä.
- Komplikaatioiden hoito. Tarttuvia komplikaatioita bakteriologisten tutkimusten tulosten saamiseksi käsitellään antibakteerisilla lääkkeillä, jotka ovat aktiivisia pneumokokkia, hemofiilisia bakteereja, meningokokkia vastaan. Käytetään penisilliiniryhmän (amoksisilliini) antibiootteja. Tromboosin kehittyessä käytetään antikoagulantteja (matalamolekyylipainoisia, fraktioimattomia hepariineja).
- Verensiirtoa. Koska anemia potilailla, joilla on kvantitatiivinen hemoglobiinipatologia, on aina vakava, hoidon perusta on säännöllinen verensiirto. Korkealaatuisia hemoglobinopatioita sairastaville ihmisille verensiirrot suoritetaan vain sekvestoinnin, hemolyyttisten ja aplastisten kriisien aikana.
- Raudan ylikuormituksen ja folaattivajeen torjuminen. Kelatoivia aineita (deferoksamiinia) käytetään ylimääräisen raudan poistamiseen kehosta. Tätä lääkettä määrätään tavallisesti yhdessä askorbiinihapon kanssa, koska se tehostaa deferoksamiinin kelatoivaa vaikutusta. Potilaiden jatkuvan hemolyysin vuoksi folaattinkulutus on lisääntynyt, joten niille osoitetaan pitkäaikainen suurten foolihapon annostus.
Kirurginen hoito
Joillekin potilaille, joilla on selvä hemolyysi, splenektoomia on tehokas hoito — pernan kirurginen poisto. Toinen kirurginen hoitotyyppi, joka mahdollistaa täydellisen remistion, on hematopoieettisten kantasolujen allotransplantaatio. Tätä menetelmää käytetään kuitenkin harvoin vain hyvin vaikeissa tapauksissa, koska se liittyy kuolemantapausten suureen esiintymistiheyteen. Kolelitiaasi sisältää kolecystectomia.
Kokeellinen hoito
Tällä hetkellä on käynnissä kliinisiä tutkimuksia hemoglobinopatioiden hoitoon. SKA-geeniterapian tulokset ovat onnistuneet. Hoidon ydin on normaalin beeta-globiiniketjun synteesiä koodaavan geenin tuominen potilaan kantasoluihin neutralisoidun lentiviruksen avulla. Lääkettä kutsutaan LentiGlobin BB305: ksi. Sen käyttö johti veriarvojen paranemiseen, mikä mahdollisti pysyvän standardihoidon hylkäämisen. Tätä lääkettä testataan myös beeta-talassemian suhteen.
Ennuste ja ennaltaehkäisy
Hemoglobinopatiat ovat vakavia sairauksia, joissa on hengenvaarallisia hengenvaarallisia komplikaatioita. Potilaat, joilla on homotsygoottinen alfa-talassemia, kuolevat ennen syntymää kohdussa. Beeta-talassemiaa sairastavat potilaat kuolevat ennen murrosikää sydämen vajaatoiminnasta. Henkilöt, joilla on korkealaatuinen hemoglobinopatia, voivat elää yli 50 vuotta asianmukaisella hoidolla. Tärkein kuolinsyy on bakteeri-infektiot, tromboottiset komplikaatiot. Useimmissa tapauksissa taudin heterotsygoottisten muotojen mukaan elinajanodote ei eroa yleisestä väestöstä.
Ensisijainen ennaltaehkäisy toteutetaan perheissä, joilla on suuri riski saada hemoglobinopatioita. Se koostuu synnytystä edeltävästä diagnoosista, jossa on abortti lääketieteellisistä merkinnöistä. Potilaiden, joilla on korkealaatuisia hemoglobiiniarvoja, on välttämättä saatava hemofilusbacillista, pneumokokista, meningokokista peräisin oleva rokote. Penisilliiniantibioottien pitkäaikainen käyttö infektioiden ehkäisemiseksi on osoitettu lapsille 4 kuukaudesta 6 vuoteen. Sama pätee potilaille, jotka saavat splenektomiaa.
Kirjallisuus
1. Anemia (A: sta Z: hen). Opas lääkäreille / Novik A.A., Bogdanov A.N. — 2004.2. Hematologinen käsikirja / Vorobyev, AI — 2005.3. Anemia (klinikka, diagnoosi, hoito) / Stuklov N. I., Alpidovsky V.K., Ogurtsov P.P. — 2013.4. Veren patofysiologia / Shiffman F.J. — 2000.
Koodi ICD-10
D58.2D56D57