Ehlers-Danlosin oireyhtymä

Ehlers Danlosin oireyhtymä, — perinnöllinen systeeminen sidekudos dysplasia, aiheuttama riittämätön kehitys kollageenin rakenteita. Riippuen kliinisestä tyyppi Ehlers-Danlosin oireyhtymä voi osoittaa hypermobility nivelet satunnaisia haavoittuvuutta ja ihon venytys, taipumus verenvuotoon ja verenvuoto, muodonmuutokset selkärangan ja kylkiluiden, myopia, karsastus, ptoosin sisäelinten ja niin edelleen. Kun diagnoosi oireyhtymä, Ehlers-Danlosin tallennettu kliininen tiedot, ihon biopsian tulokset ja genotyypitys; Perinnöllisen patologian diagnoosi on mahdollista. Oireyhtymän hoitaminen, Ehlers-Danlosin syndrooma rajoittuu noudattaminen lempeä hoitoon, proteiini ruokavalio, oireenmukainen hoito.
- Ehlers-Danlos-oireyhtymän syyt
- Ehlers-Danlosin oireyhtymän luokittelu
- Ehlers-Danlos-oireyhtymän oireet
- Ehlers-Danlos-oireyhtymän diagnosointi
- Ehlers-Danlos-oireyhtymän hoito
- Ehlers-Danlosin oireyhtymän ennuste
Ehlers-Danlosin oireyhtymä
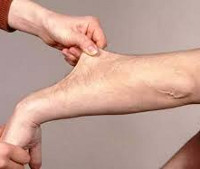
Ehlers-Danlosin oireyhtymä (puutteellinen desmogenez, giperelasticheskaya iho) yhdessä osteogenesis imperfecta Marfanin oireyhtymä ja muut sairaudet, jotka liittyvät perinnöllinen kollagenopatiyam. Ehlers-Danlosin oireyhtymä on heterogeeninen ja sisältää heterogeeninen joukko perinnöllisiä sidekudoksen leesioita (dysplasiat sidekudos), johon liittyy heikentynyt biosynteesiä kollageeni. Ilmenemismuotoja Ehlers-Danlosin oireyhtymä ovat systeemisiä luonteeltaan ja vaikuttavat tuki- ja liikuntaelimistön, iho, sydän, visuaalinen, hampaiston, ja muita järjestelmiä. Näin ollen, Ehlers-Danlosin oireyhtymä on käytännön merkitystä paitsi genetiikan vaan myös traumatologiaan ja ortopedian, ihotaudit, kardiologian, silmätaudit, hammaslääketieteen.
Tarkastuksen monimutkaisuus ja valomuotojen esiintyminen vaikeuttavat tarkkoja tietoja Ehlers-Danlosin oireyhtymän todellisesta esiintyvyydestä. keskimäärin 1: 5 000 vastasyntynyt, raskas muoto — 1: 100 000.
Ehlers-Danlos-oireyhtymän syyt
Ehlers-Danlosin oireyhtymän erilaiset variantit eroavat perinnöllisyystyypistä, ensisijaisista molekyyli- ja biokemiallisista virheistä. Kaikissa kliinisissä muodoissa on kuitenkin sellaisten geenien mutaatioita, jotka aiheuttavat kollageenin kvantitatiivista tai rakenteellista patologiaa. Tähän mennessä Ehlers-Danlosin oireyhtymän molekyylimekanismeja ei ole todettu sairauden kaikista muodoista.
Näin ollen on tunnettua, että tyypin I oireyhtymä ominaista alentunut aktiivisuus fibroblastien, lisääntynyt synteesi proteoglykaanien puute vastaavien entsyymien normaalia biosynteesiä kollageenin. Ehlers-Danlos-tyypin IV oireyhtymä liittyy tyypin III kollageenin tuotannon riittämättömyyteen; VI tyypin tauti esiintyy vajaatoiminnan lysyyli hydroksylaasin entsyymi, joka osallistuu hydroksylaation lysiinin prokollageenin molekyylejä. VII-tyyppi johtuu siitä, että Procollagen-tyypin I muuttaminen kollageeniksi on rikottu; X-tyyppi — plasman fibronektiinin patologia, joka liittyy solunsisäisen matriisin organisointiin jne.
Patologinen malli eri Ehlers-Danlosin oireyhtymä tunnettu siitä, ohenemista dermis, sekavuus, ja menetys tiiviyttä kollageenisäikeiden, kasvua elastisia kuituja, kasvaa alusten lukumäärä ja Ontelon laajentuminen.
Ehlers-Danlosin oireyhtymän luokittelu
Yhteensä on 10 erilaista Ehlers-Danlos-oireyhtymää, joka poikkeaa geneettisestä puutteesta, perinnöstä ja kliinisistä ilmenemismuodoista. Harkitse tärkeimmät:
I-tyypin Ehlers-Danlos -oireyhtymä (klassinen raskas sairaus) on taudin yleisimpiä variantteja (43% tapauksista), joilla on autosomaalinen hallitseva perinnöllisyys. Varsinainen oire on ihon hyperelasticiteetti, jonka laajeneminen on 2-2,5 kertaa normaalia korkeampi. Luonnossa yleistyneiden nivelten hypermobility, luuston muodonmuutokset, lisääntynyt ihon heikkous, taipumus ulkoiseen verenvuotoon, arven muodostuminen, haavojen heikko paraneminen on ominaista. Joillakin potilailla ilmenee nilviäisten kaltaisten pseudotumorien ja alemman ääripäiden suonikohjujen esiintyminen. Raskaus naisilla, joilla on tyypin I Ehlers-Danlos-oireyhtymä, on usein monimutkainen ennenaikaisen syntymän varalta.
II-tyyppinen Ehlers-Danlosin oireyhtymä (klassinen pehmeä virtaus), jolle on tunnusomaista edellä kuvatut merkit mutta ilmaistu pienemmäksi. Ihon venytys ylittää normaalin vain 30%; hypermobility havaitaan pääasiassa jalkojen ja käsien nivelissä; verenvuoto ja arpeutuminen ovat merkityksettömiä.
III-tyyppinen Ehlers-Danlos-oireyhtymä — on autosomaalinen hallitseva perintö, hyvänlaatuinen kurssi. Kliinisiin ilmiöihin sisältyy yleistynyt nivelten liikkuvuus, tuki- ja liikuntaelinten epämuodostumat. Muut oireet (ihon hyperelasticisuus ja arpeutuminen, verenvuodot) ovat vähäisiä.
IV-tyypin Ehlers-Danlos-oireyhtymä — on harvinaista, se on vaikeaa; voidaan periä eri tavoin (hallitseva tai recessive). Ihon hyperelasticiteetti on vähäpätöinen, vain sormien liitosten liikkuvuus lisääntyy. Tämän tyyppisen taudin johtava ilmentymä on verenvuotoinen oireyhtymä: taipumus ekmimoosin muodostumiseen, spontaani hematooma (mukaan lukien sisäelimet), onttojen elinten ja alusten repeytyminen (mukaan lukien aortta). Sen mukana seuraa suuri kuolemantapaus.
V-tyypin Ehlers-Danlos-oireyhtymää — on X-sidottu resessiivinen perintönä. Sille on ominaista lisääntynyt ihonsiirtyvyys, kohtuullisesti ilmaantunut nivelten hypermobility, verenvuoto ja ihon herkkyys.
VI-tyypin Ehlers-Danlos-oireyhtymä — perinnöllinen autosomaalinen recessive tyyppi. Ihon hyperelasticisuuden, verenvuodon kaltevuuden ja nivelten liikkuvuuden lisäämisen lisäksi on lihasrelpotus, vaikea kyphoscoliosis, clubfoot. Ehlers-Danlosin tyypin VI oireyhtymän ominaispiirre on silmäsairaus, jota ilmenee myopiassa, keratoconuksessa, strabismuksessa, glaukooma, verkkokalvon irtoamisessa jne.
VII-tyyppinen Ehlers-Danlos-oireyhtymä (arthroclasia) — periytyy autosomaalisena dominanttina ja autosomaalisena resessiivisenä. Kliininen kuva määräytyy potilaiden vähäisen kasvun ja nivelten hyperplastisuuden vuoksi, mikä johtaa usein toistuviin poikkeamiin.
VIII-tyyppinen Ehlers-Danlos-oireyhtymä — pääasiassa perinnöllinen autosomaalinen määräävä. Johtava rooli klinikassa on ihon heikkous, voimakas periodontiitti, joka johtaa varhaisen hampaan menetykseen.
X-tyyppinen Ehlers-Danlos-oireyhtymä — karakterisoitu autosomaalisella resessiivisellä perinnöllä; ihon kohtalainen hyperelasticiteetti ja nivelten hypermobility, striae (ihon raidallinen atrofia), verihiutaleiden aggregaation rikkominen.
XI-tyyppinen Ehlers-Danlos-oireyhtymä — on autosomaalinen hallitseva perintötyyppi. Potilaita, joilla on toistuva harvennusliitosten sumentuminen, patelarajakytkentöjä, synnynnäistä lonkan poikkeamista esiintyy.
IX-tyyppinen (X-taitettu verilöylyversio) on tällä hetkellä poissuljettu Ehlers-Danlos-oireyhtymän luokittelusta. Ehlers-Danlosin oireyhtymän nykyaikaisessa versiossa on 7 pääasiallista sairaustyyppiä:
- Klassinen (tyypit I ja II)
- hypermobiili (tyyppi III)
- verisuonisto (tyyppi IV)
- kyphoscoliosis (tyyppi VI)
- arthroclasia (tyyppi VIIB)
- dermosparaxis (tyyppi VIIC)
- tenascin-X: n puuttuminen
Ehlers-Danlos-oireyhtymän oireet
Ottaen huomioon, että Ehlers-Danlosin oireyhtymän yksityiskohtainen kuvaus annetaan edellä, tässä osiossa esitetään yhteenveto taudin tärkeimmistä ilmenemismuodoista. Koska sidekudos on läsnä lähes kaikilla elimillä, Ehlers-Danlosin oireyhtymän ilmentymät ovat systeemisiä, yleistettyjä luonnossa.
Johtava kliininen kuva on iho-oireyhtymä: ihon hyperelasticiteetti, joka kerätään helposti kerralla ja vedetään takaisin. Ihon kosketusnäyttö on samettinen, herkkä, hieman kiinnittynyt taustalla olevien kudosten kanssa, rypistynyt palmari- ja istutuspintoihin. Ehlers-Danlosin oireyhtymän ihon hyperelasticiteetti on havaittu syntymän tai esikouluajan jälkeen, ja se on vähentynyt vuosien varrella.
Lisäksi hyper-laajennettavuus on ominaista lisääntynyt haavoittuvuus, ihon haurastuminen, joka näkyy yli 2-3 vuoden ikäisenä. Minimaalinen trauma johtaa pitkän aikavälin paranemishaavojen muodostumiseen, jonka paikalleen jonkin aikaa on muodostunut atrofisia tai keloidisia arpia pseudotumoraaleja.
Ehlers-Danlosin oireyhtymän yhteisiä ilmenemismuotoja edustavat liitosten hypermobility (laajentuminen), jotka voivat olla paikallisia (esimerkiksi interfaargaalisten nivelten uudelleen avaaminen) tai yleistynyt luonne. Artikulaarinen oireyhtymä ilmaantuu lapsen kävelyn puhkeamisen myötä, mikä johtaa toistuviin subluksointeihin ja hajoamiseen. Iän myötä nivelen hypermobility vähenee yleensä.
Sydän- ja verisuonijärjestelmästä lapsilla, joilla on Ehlers-Danlos-oireyhtymä, on usein synnynnäisiä sydämen vajaatoimintoja, mitraalisten venttiilien prolapsia, aivoverenvuotoja, suonikohjuja. On olemassa taipumusta verenvuotoon — kouristukset, erilaisten paikallistumien hematooma, nenän, gingivaalit, kohdun, ruuansulatuskanavan verenvuodot.
Silmäoireita Ehlers-Danlosin oireyhtymä voi sisältää giperelastichnost silmäluomen iholle, myopia, ptoosia, karsastus, sarveiskalvon kyyneleet ja silmämunan minimaalisella mekaanisia vaurioita, spontaani verkkokalvon irtauma.
Muutokset luuranko Ehlers-Danlosin oireyhtymä tunnettu siitä, että suppilon tai keeled muodonmuutos rintakehän, skolioosi, kyphosis, kampurajalka, ylipurenta, osittain hampaaton. Sisäelinten häiriöiden esitetty ptosis sisäelinten, navan, imusolmukkeet, palleatyrä, toistuvat spontaanit ilmarinta, suoliston divertikuloosi et ai. Mental kehittäminen lasten Ehlers-Danlosin syndrooma vastaa yleensä ikää.
Ehlers-Danlos-oireyhtymän diagnosointi
Diagnoosi Ehlers-Danlosin syndrooma pidetään lääketieteellisen genetiikan perustuvat sukututkimustietoja, sairaushistoria, kliiniset tutkimukset, molekyyligenetiikan tutkimukset. Pre-Ehlers-Danlosin syndrooma voidaan epäillä läsnäollessa suurten diagnostisten kriteerien (yhteinen hypermobility, giperelastichnosti iho, taipumus verenvuotoon) ja hyvin pieni (ihon haurauteen, sydänsairaudet, verisuonten, silmien, ja niin edelleen. D.).
Jotkut taudin muodot edellyttävät ihon biopsiaa histologista, histokemiallista, elektronimikroskooppista tutkimusta varten.
Ehlers-Danlosin oireyhtymän potilaiden läsnäolo on osoitus lääketieteellisestä geneettisestä neuvonnasta ja invasiivisesta prenataalisesta diagnoosista.
Potilaat, joilla on erilaisia Ehlers-Danlosin oireyhtymä voi vaatia tarkkailu ja tutkiminen lasten ortopedisen vamman, lasten kardiologi, lasten silmälääkäri, lasten hammaslääkäri, verisuonten kirurgi.
Ehlers-Danlos-oireyhtymän hoito
Ehlers-Danlosin oireyhtymän tehokasta erityishoitoa ei ole kehitetty. Lapset tarvitsevat säästää hoitoa, joka sulkee pois liitosten ja ihon tarpeeton traumatisaation; liikunnan rajoittaminen; sitoutuminen proteiinin ruokavalioon sisällyttämällä ruokavalioon luun liemet, hyytelöt, hyytelö. Säännöllinen hieronta, fysioterapia, fysioterapia (magneettihoito, elektroforeesi, laserpunktio) ovat pakollisia.
Huumeiden oireyhtymä, Ehlers-Danlosin terapiassa käyttöä aminohappojen (karnitiinin), vitamiinit (C, E, D, B-ryhmän), kondroitiinisulfaatti, glukosamiini, kivennäisaineita (kalsium ja magnesium), metaboliset aineet (Riboxinum, ATP, koentsyymi Q10) toistetaan kursseja jopa 1-1,5 kuukautta. 2-3 kertaa vuodessa.
Kun Ehlers-Danlosin oireyhtymä, voidaan osoittaa leikkauksen: rekonstruktio rintakehän, poistamalla pseudotumor UPU korjaus ja niin edelleen.
Ehlers-Danlosin oireyhtymän ennuste
Ehlers-Danlosin oireyhtymää sairastavien potilaiden laatu ja kesto vaikuttavat taudin tyyppiin. Vakavin ennuste on Ehlers-Danlosin oireyhtymä IV-tyyppi — kuolemaan johtava tapaus voi ilmetä alusten, sisäisten elinten ja verenvuodon puhkeamisen seurauksena. I-tyypin oireyhtymän esiintyminen rajoittaa merkittävästi elämänlaatua. Suhteellisen edullinen II-III -tyyppisten tautien kulku.
Yleensä Ehlers-Danlos-oireyhtymän läsnäolo on täynnä sosiaalisia vaikeuksia, rajoittaa koko liikuntaa ja ammatin valintaa.