Craniometaphyseal dysplasia
Kraniometafizarnaya dysplasia — perinnöllinen tila ryhmästä, osteokondrodysplasioiden tunnettu siitä, epämuodostumia kallon ja raajojen luita metafyysien. Oireita tätä tautia ovat hypertelorismi, epämuodostumia kasvojen usein vahingoita potilaan epänormaali muodostuminen Nenäkäytävien loukkaa heidän aukioloa, päänsäryt ja kaartuu raajat. Diagnoosi dysplasia kraniometafizarnoy suorittaa perusteella tutkimuksen esillä olevan potilaan tila, X-ray tiedot ja tulokset molekyyligeneettisten tutkimuksia. Erityistä hoitoa tähän sairauteen ei ole olemassa, käytä oireenmukainen hoito joissakin tapauksissa leikkaus suoritetaan lievittää potilaan hengitys ja parantaa sen ulkonäköä.
- Craniometaphyseal dysplasian syyt
- Craniometaphyseal dysplasian oireet
- Craniometaphyseal dysplasian diagnosointi ja hoito
Kraniometalfysikaalinen dysplasia
Kraniometafizarnaya dysplasia — joukko perinnöllisiä sairauksia, jotka johtavat epämuodostuma kasvojen luut kallo ja aivoleikkeille lievempiä yhdessä anomalia pitkien luiden metafyysialueilla ja muut häiriöt. Aikaisemmin tämä tila viittaa samaan ryhmään taudin Pylen (useita Metafysiaalinen dysplasia, dysplasia kraniometafizarnaya Pyle), mutta se on nyt eristetty erillisessä tauti yksiköitä. Tämä johtuu siitä, että tämä sairaus on yleinen ovat kallon rakenne poikkeavuuksia (muodonmuutos hyperostoo- ja luun skleroosi), kun taas patologian muut osat luuranko ilmaistuna heikosti. On olemassa kahta perustyyppiä kraniometafizarnoy dysplasia, eroavat mekanismi perintö (autosomaalinen dominoiva ja peittyvästi tyypit) kliiniset oireet ja sairauksien vakavuus. Autosomien kautta tapahtuvan perinnöllisyyden ansiosta yhtä todennäköinen patologia vaikuttaa sekä miehiin että naisiin. Kraniometaphyseaalisen dysplasian esiintymistiheyttä ei ole määritetty tarkasti, hallitseva tyyppi on usein useammin kuin recessive-tyyppinen.
Craniometaphyseal dysplasian syyt
Pääasiallinen syy yleisempiin, mutta helpompiin, autosomaaliseen hallitsevaan muotoon kraniometaphyseaalinen dysplasia ovat mutaatioita ANKH-geenissä, joka sijaitsee viidennellä kromosomilla. Geniini koodaa proteiinia, joka on pyrofosfaatin membraani kantaja, joka osallistuu luukudoksen kalkinpoistomenetelmien ja sen resorption inhibointiin. Seurauksena on geenivirhe siirtävän proteiinin rakenne muuttuu, ja se ei kykene täysin täyttää tehtäväänsä, mikä johtaa kraniometafizarnoy dysplasia. Solutasolla, tämä ilmenee epänormaali muutokset osteoklastien toimintaa, kehittää tauti ja hyperostoosin lähinnä luut kallo. Myös kallon pohjan pääreikien kokoonpano, mikä aiheuttaa joidenkin alusten ja hermojen puristusta ja määrittää suurelta osin loput kraniometaphaseaalisen dysplasian oireet (kuulon menetykset, päänsäryt, trigeminaalin ja kasvojen hermojen tappion). Geenitekniikassa tunnetaan muita sairauksia, jotka johtuvat ANKH-geenin tukahduttamisesta, erityisesti — perinnöllinen pseudogout tai perimän chondrocalcinosis.
Joka on paljon harvinaisempi peittyvästi periytyvä muoto kraniometafizarnoy dysplasiaa johtuu viasta GJA1 geeni kromosomissa 7 minuuttia. Sen ilmentyminen on proteiini nimeltään konneksiini 43, osallistuu aktiivisesti muodostumiseen solujen välisen (mezhschelevyh) koskettaa monissa kudoksissa, joka sallii solujen vaihdon matalan molekyylipainon yhdisteitä. Patogeneesin dysplasian kraniometafizarnoy missensemutaatioita c716G> A on tiedossa, suurin ongelma on pienentynyt syiden tunnistaminen eristetyn vaurioiden kallon luut, ja metaphyseal luustomuutoksia suhteellisen ilman muihin elimiin. Koska eniten konneksiini 43 ihmisellä on sydänkudoksen kysymys puute sydänsairauksien tiettynä mutaatio mennessä on mysteeri useimmille lääkäreille, geneetikkojen. vain
Craniometaphyseal dysplasian oireet
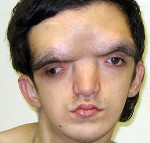
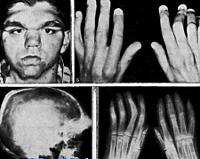
Automaattinen dominoiva tyypin kraniometyypin dysplasia on tyypillistä kevyemmällä kurssilla, usein lapsen syntymällä ei havaita taudin oireita. Ainoastaan ensimmäisellä elämänvuodella on hypertelorismi nenäsillan laajentumisen vuoksi nenän luiden hyperostosiasta. Tulevaisuudessa kraniometyypin dysplasia johtaa nenän limakalvon kaventumiseen ja niiden läpäisevyyden rikkomiseen, joten potilaat avaavat usein suunsa, koska nenän hengitys heikkenee. 6-7-vuotiaana voidaan alkaa määritellä pitkät putkimaiset luut, jotka ulkonäöltään ilmenevät polvien ja kyynärliitosten koon kasvaessa. Noin puolet potilaista, joilla on kraniometaphyseaalinen dysplasia, kehittävät kuulovammaisia, joilla on erisuuruinen kokonaiskuurosumma — useimmiten tämä johtuu luukadon kuulohäiriön puristumisesta.
Kraniometafizarnaya dysplasia peittyvästi periytyvä tyypin on tunnusomaista paljon vaikeita epämuodostumia kallon luut ja raajat, merkkejä patologiasta havaitaan usein heti syntymän jälkeen. Potilailla esiintyy merkitty hypertelorismi, kasvonpiirteet ovat usein erittäin epäsymmetrinen, muodonmuutos nenän kuonon ja voi hankkia merkkejä epämuodostuma. Joissakin tapauksissa on olemassa macrocephaly, leuanalussylkirauhasista alapurenta ja muut malocclusion ja hampaiston. Potilaan kehittymisen myötä kallon luiden epämuodostumat voivat pahentua. Metafyysien raajojen parannettu huomattavasti, mikä usein aiheuttaa toissijainen muodonmuutoksen (esim., X-muotoinen kaarevuus jalkaa). Kuten edellisessä suoritusmuodossa, tässä muodossa kraniometafizarnoy dysplasia on usein erilaisia neurologisia häiriöitä, jotka johtuvat aivojen hermojen puristumisesta ja traumasta. Ne voivat ilmetä kuuroina, näkövammaisina, kasvojen ihon herkistymisen häiriöissä, kasvojen lihasten ja päänsärkyjen paresisissa. On joitakin kuvauksia potilaista samanaikaisesti kärsivien kraniometafizarnoy dysplasia ja kehitysvammaisuus, mutta luotettavaa tietoa suhteesta näiden kahden valtion tähän mennessä ei.
Craniometaphyseal dysplasian diagnosointi ja hoito
Craniometaphyseal dysplasian diagnoosi perustuu potilaan tutkimustietoon, perinnöllisen historian tutkimukseen, röntgentutkimusten tuloksiin ja molekyyligeneettisiin analyyseihin. Tutkittaessa määritetään erilaisia epämuodostumia kallon luiden kehittymisessä, mikä heijastuu potilaan kasvojen piirteisiin. Tässä tapauksessa autosomaalinen hallitseva muoto, erityisesti pikkulapsissa, osoittaa itsensä melko heikosti suhteessa tähän oireeseen. Molemmissa kraniometyylipatulehduksen tyypissä hypertelorismi, nenäsillan laajentuminen nenän luiden kasvusta johtuen poskipyöriä kohti, nenän limakalvon kaventuminen tai tukkeutuminen ovat lähes aina läsnä. Fyysisen tutkimuksen ja lisätutkimusten puitteissa neurologisia häiriöitä voidaan myös havaita: kuulon väheneminen tai puuttuminen, näkökyvyn väheneminen,
Paljon tietoa kraniometafizarnoy dysplasiaa tarjoavat radiologisen tutkimusmenetelmiä luuranko. X-ray kallon autosomaalinen dominantti muodossa tauti määritetään tiivistys luukudoksen alueella niskakyhmyä kallonpohjan skleroosi, vähentää pneumatization sinin ja solujen temporaaliluualueen. Joissakin tapauksissa, voidaan tunnistaa skleroosi intercostals nivelet ja laajeneminen pitkien luiden metafyysialueilla. Autosomisesti-resessiivinen tyyppi kraniometafizarnoy dysplasia röntgenkuvissa paljasti samanlainen, mutta paljon enemmän vakavia sairauksia, kuten — täydellinen puuttuminen nenän sivuonteloiden, ja joskus terävä kaventuminen luun täyttöaukot kallohermojen. Lisäksi on tauti, ei vain pohja, mutta myös kallon holvin, joissakin tapauksissa merkittävästi muodonmuutos kasvojen osan luun.
Perinnöllisen historian ja geneettisen diagnoosin tutkimusta käytetään myös aktiivisesti kraniometaphyseaalisen dysplasian määrittämiseen. Tässä tapauksessa voit ensin määrittää taudin perinnöllisen lajin, jonka avulla voit säätää molekyyligeneettistä analyysia mutaatioiden löytämiseksi tietyllä geenillä. Tätä varten käytetään ANKH- ja GJA1-geenien suoraa sekvensointimenetelmää.
Erityistä hoitoa kraniometafizarnoy dysplasiaa ei ole olemassa, määrätä oireenmukainen hoito, johon liittyy erilaisia kirurgisia tekniikoita. Viimeksi mainittu, erityisesti, voi parantaa läpäisevyyttä nenäkäytävien helpottaa hengitystä. Lievittää neurologisia oireita voidaan saavuttaa laajentamalla vastaaviin aukkoihin kallohermojen. Lisäksi muovi kirurgit pystyvät minimoimaan vakavuus esteettisiä vikoja kraniometafizarnoy dysplasia. Parantaa kuulemistilaisuus kuulolaitteita.
Craniometaphyseal dysplasian ennuste ja ehkäisy
Monissa tapauksissa, ennuste kraniometafizarnoy dysplasian autosomaalinen hallitseva tyyppi suhteessa potilaan selviytymisen suotuisa — ei anomalia kallon tai toissijainen neurologisia häiriöitä eivät johtaa vakaviin seurauksiin. Lapsuudessa syntyvien kuulon tai näkökyvyn sairaudet voivat hitaasti kehittyä 20-30 vuoteen, minkä jälkeen huononeminen ei tavallisesti tapahdu. Peittyvästi kraniometafizarnoy dysplasiaa on tunnusomaista huono ennuste, koska kallon luusto tämän ehdon taipumus pahentaa ja ovat joka vuosi enemmän ja enemmän vahingoittaa hermo. Joissakin tapauksissa tämä johtaa autonomisen hermoston osaksi halvaantumista ja turhautumista. Craniometaphyseal-dysplasian ehkäisyä ei ole kehitetty,