Atelosteogenez

Atelosteogenesis on geneettisten patologioiden ryhmä — chondrodysplasia, joka usein johtaa kuolemaan johtaneeseen lopputulokseen varhaisessa iässä tai kohdunsisäisen kehityksen vaiheessa. Tärkeimpien oireiden sairauksien työntyvät hypoplasia reisiluun ja olkaluun luut niiden distaalisen lyhentäen ripojen ja kaventuminen rintakehän, dislokaatiot nivelten ja kaarevuus putkimaisen luita. Atelosteogeneesin diagnoosi suoritetaan röntgenkuvan perusteella luurakenteista, potilaan nykyisestä tilasta ja geneettisten tutkimusten tuloksista. Tällä hetkellä atelosteogeneesin muotoa ei ole olemassa.
- Atelosteogeneesin syyt
- Atelosteogeneesin luokitus
- Atelosteogeneesin oireet
- Atelosteogeneesin diagnosointi ja hoito
Atelosteogenez
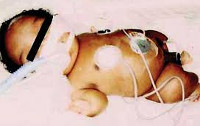
Atelosteogenesis (de la Chapellen dysplasia, vastasyntyneiden luun dysplasia) on tappavan chondrodysplasian muoto, jolle on ominaista korkea kuolleisuus varhaisessa iässä. Yleisin patologian muoto (tyypin II atelosteogenesis) periytyy autosomisen recessive-mekanismin avulla. Tyyppien 1 ja 3 tyypit johtuvat autosomaalisen hallitsevan tyypin perimäisistä mutaatioista, mutta tällaisen atelosteogeneesin yleisin syy on geneettinen vaurio de novo. Tapahtuma on melko alhainen — ei enempää kuin 1: 1 000 000, samalla todennäköisyydellä sekä poikien että tyttöjen sairaus. On olemassa yksittäisiä tapauksia, joissa potilaat selviytyivät aikuisiän tai edenneen iän, valtaosa heistä kärsii tyypin 2 aterogeneesin ja vain yhden tyypin 3 sairaudesta.
Atelosteogeneesin syyt
Atelosteogeneesin syy ovat FLNB-geenien (1. ja 3. tyypit) ja SLC26A2: n (tyyppi 2) mutaatiot. Geeni FLNB, joka sijaitsee kolmannessa kromosomissa, joka koodaa proteiinia kutsutaan filamiini B, joka osallistuu rakentamiseen solunsisäisen runkorakenteen sisältyy lukuisia sidekudosta. Muodot atelosteogeneza aiheuttama vaurio geenin tunnettu siitä, autosomaalinen dominantti perintönä, mutta koska suuri kuolleisuus tuollaisista mutaatioista ovat yleisempiä spontaanin lajeja. Häiriön takia proteiinin rakenteen filamina esiintyä useita luuston epämuodostumia, sidekudoksen, sisäelimiin, mikä usein johtaa sikiön kuolemaan tai kuolema varhaisessa iässä.
Tyypin 2 atelosteogenesis, joka aiheutuu SLC26A2-geenin mutaatiosta, on ominaista autosomaalisella resessiivisellä perintötyypillä. Tämä geeni sijaitsee kromosomissa 5, koodaa kuljettajaproteiinin rakenne sulfaatti-ioneja, on keskeinen rooli muodostumista ruston proteoglykaanien. Myös mukana muodostumista rustonsisäisen luun erilaisia, joten vastaisesti proteiinin rakenteiden näkyvät epämuodostumat, tuki- ja liikuntaelinten, ominaisuus atelosteogeneza. Lisäksi erilaiset poikkeavuudet sisäelinten muodostumisessa ovat mahdollisia — keuhko-hypoplasia, synnynnäiset sydämen vajaatoiminta. Kaikki tämä johtaa siihen tosiasiaan, että tyypin II atelosteogenesis-potilaat usein kuolevat varhaisessa iässä.
Atelosteogeneesin luokitus
Tällä hetkellä on kolme pääasiallista tautityyppiä:
Tyypin 1 atelosteogenesis johtuu FLNB-geenin letaalisesta mutaatiosta, joka useimmissa tapauksissa esiintyy de novo. Se on vakavin kehityshäiriö, joka johtaa monissa tapauksissa sikiön kuolemaan ennen synnytystä.
Atelosteogeenityyppi 2 (dysplasia de la Chapelle) aiheutuu SLC26A2-geenin mutaatiosta, joka välitetään autosomaalisen recessivismekanismin kautta. Ennenaalinen tai perinataalinen kuolema tällä lomakkeella on harvinaista, useimmissa tapauksissa potilaat kuolevat ensimmäisinä kuukausina tai elinvuosina sisäisten elinten toimintahäiriöistä.
Tyypin 3 atelosteogenesis johtuu myös mutaatiosta FLNB-geenissä, joka välitetään autosomaalisella dominanttimekanismilla tai tapahtuu spontaanisti. Kuolema tulee yleensä ensimmäisten elinviikkojen aikana tiettyjen elinten vaikeasta kehittymisestä.
Useimmissa tapauksissa neonatologin, joka perustuu vain potilaan tutkimustietoihin ja röntgenkuvaan, ei pysty erottamaan yksittäisiä atelosteogeneesiä. Geenitekniikan voi tehdä vain sekvensoimalla sairauksia liittyvien geenien sekvenssejä ja määrittämällä mutaation tyyppi.
Atelosteogeneesin oireet
Taudin oireille on ominaista tietty vaihtelu. Useimmissa tapauksissa potilailla, joilla on vastasyntyneiden havaittu lyhentäminen ylä- ja alaraajojen koko pienenee rinnassa, takaisinvedon peukalo ja varpaat. Kasvot tasainen, todetaan selvästi hypertelorismi, takaisinvedon nenän ja takana nenän. Vastasyntyneet paljasti myös hypoplasia keuhkokudoksessa, joilla on usein sydänsairaus tai ahtauma kurkunpään, jotka usein ovat kuolinsyy. Jos atelosteogenezom potilaista kuolee ensimmäisinä päivinä ja viikkoina elämän, sitten ne diagnosoidaan lausutaan skolioosi, equinovarus jalka epämuodostuma, kampurajalka. Kuolleisuus vanhemmassa iässä johtuu myös samanaikaisista sisäelinten rikkomuksista.
Atelosteogeneesin diagnosointi ja hoito
Atelosteogeneesin diagnosointi tehdään potilaan nykyisen tilan, radiografisten tutkimustietojen ja nykyaikaisten genetiikan menetelmien perusteella. Samanaikaisesti röntgensäde näyttää pitkät putkimaiset luut kaarevuuteen, koska niiden etäisyyksillä ei ole luumutusta. Rintaelinten röntgenkuvat paljastavat ristin luutumisen lyhentämisen ja rikkomuksen, rintakehän muodonmuutoksen ja ruumiiden leviämisen ruumiissa. Tutkimuksen aikana löydetään usein polven, lonkan ja lantion liitokset. Luun luiden värjäytyminen on rikki.
Geneettiset menetelmät atelosteogeneesin diagnoosiksi pelkistetään sekvensoimalla FLNB- ja SLC26A2-geenien sekvenssi mutaatiotyypin tunnistamiseksi ja määrittämiseksi. Differentiaalinen diagnoosi olisi tehtävä muiden perinnöllisten vastasyntyneiden dysplasiaa (dyspepsia, Larsenin oireyhtymä). Myös atelosteogeneesin synnytyksen diagnoosi on mahdollista ekografisten tai geneettisten (amniocentesis) menetelmien avulla.
Tällä hetkellä ei ole erityistä etiotrooppista hoitoa minkään atelosteogeneesin muodolle. Tukihoito vähenee elvytettävästi synnytyksen jälkeisiin interventioihin, joissakin tapauksissa — kirurgisia toimenpiteitä sydämen vajaatoiminnan tai muiden sisäisten häiriöiden korjaamiseksi. Ennuste on erittäin epäsuotuisa, vain muutamat prosentit atelosteogeneesiä sairastavista potilaista selviävät vuoden ikäisiksi, kertoivat yksittäiset tapaukset, joissa potilaat elivät aikuisikään.