Amyloidoosi
Amyloidoosi — yhteinen systeeminen sairaus kehon jossa kerrostumista tietyn glykoproteiinin (amyloidi) elinten ja kudosten kanssa heikentyneen toiminnan jälkimmäisen. Kun amyloidoosi voi vaikuttaa munuaisten (nefroottinen oireyhtymä, turvotus oireyhtymä), sydämen (sydämen vajaatoiminta, rytmihäiriöt), maha, tuki- ja liikuntaelimistön, ihoa. Mahdollinen polyserositiitin, hemorrhagisen oireyhtymän kehitys, mielenterveyden häiriöt. Amyloidoosin luotettavaa diagnoosia helpottaa amyloidin havaitseminen vaikuttavien kudosten biopsia-näytteissä. Immunosuppressiivista ja oireista hoitoa käytetään amyloidoosin hoitoon; indikaatioista — peritoneaalidialyysi, munuaisten ja maksansiirto.
- Amyloidoosin syyt
- Amyloidoosin luokitus
- Amyloidoosin oireet
- Amyloidoosin diagnoosi
- Amyloidoosin hoito
amyloidoosi
Amyloidoosi — tauti ryhmästä, systeeminen disproteinozov virtaavan muodostumista ja kertymistä kudoksiin proteiini-polysakkaridi kompleksiyhdisteen — amyloidin. Esiintyvyys amyloidoosin maailmassa pitkälti maantieteellisesti määritelty: esimerkiksi määräajoin tauti on yleisempää maissa Välimeren altaaseen amyloidipolyneuropatiaa — .. Japanissa, Italiassa, Ruotsissa, Portugalissa, jne keskimääräinen tiheys amyloidoosin väestöstä on 1 tapaus 50000 väestöstä .. Amyloidoosi kehittyy yleensä yli 50-60-vuotiailla ihmisillä. Koska kun amyloidoosi koskee lähes kaikkia elinjärjestelmiin, tauti tutkineet erilaisia lääketieteellisiä hoitomuotoja: reumatologia, urologian, kardiologian, gastroenterologian, neurologian ja muut.
Amyloidoosin syyt
Etiologia ensisijainen amyloidoosin ei ole täysin ymmärretty. Kuitenkin, se on tunnettua, että toissijainen amyloidoosi liittyy tavallisesti krooninen tarttuva (tuberkuloosi, syfilis, aktinomykoosi, malaria) ja pyo-tulehdussairauksien (osteomyeliitti, bronkiektasiatauti, keuhkopussin empyema, bakteeriendokardiitti, jne.), Ainakin — neoplastisiin prosesseihin (Hodgkin , leukemia, munuaissyöpä ja keuhkosyöpä). Reaktiivinen amyloidoosi voi esiintyä potilailla, joilla ateroskleroosi, nivelreuma, selkärankareuma, psoriaasi, haavainen paksusuolen tulehdus, Crohnin tauti ja Whipple sarkoidoosi.
Se havaitsi erilaisia muotoja perinnöllinen amyloidoosi: Välimeren kuume (jaksoja kuume, vatsakipu, ummetus, ripuli, keuhkopussintulehduksesta, niveltulehdus, ihottuma iholla), Portugali neuropaattinen amyloidoosi (perifeerinen polyneuropatia, impotenssi, sydämen johtuminen), Suomen tyyppi (surkastuminen sarveiskalvo, kallon neuropatia), Tanskan versio (kardiopatichesky amyloidoosi) ja monet muut. et ai. Tekijöitä, edesauttamaan amyloidoosin, hyperglobulinemia ensiarvoisen tärkeää, toimintahäiriö soluimmuniteetin, geneettinen alttius, ja muut.
Amyloidoosin patogeneesi
Joukossa monia versioita amiloidogeneza eniten kannattajia on disproteinoza teoria, paikalliset solu synty, immunologisia, ja mutaatio teoria. Teoria paikallisen solun synnystä katsoo vain prosessoi esiintyvät solutasolla (muodostuminen säikeisen amyloidiprekursoriproteiinin makrofagi-järjestelmän), kun taas muodostumista ja kertymistä amyloidin tapahtuu solujen ulkopuolella. Tästä syystä paikallisen solugeesin teoriaa ei voida pitää tyhjentäväinä.
Disproteinoosin teorian mukaan amyloidi on epänormaalin proteiinimateriaalin metaboliaa. Tärkeimmät patogeneesi amyloidoosin — Dysproteinemia hyperfibrinogenemia ja edistää kertymistä plasmaan proteiinifraktiot ja karkea paraprotein.
Amyloidoosin alkuperää oleva immunologinen teoria sitoo amyloidin muodostumisen antigeenin vasta-ainereaktiolla, jossa vieraat proteiinit tai sisäisten kudosten hajoamistuotteet toimivat antigeeneinä. Tässä tapauksessa amyloidin kertyminen tapahtuu pääasiassa vasta-aineiden muodostumispaikoissa ja ylimääräisissä antigeeneissä.
Yleisimmistä on amyloidoosin mutaatioteoria, jossa otetaan huomioon valtava valikoima mutageenisiä tekijöitä, jotka voivat aiheuttaa epänormaalia proteiinisynteesiä.
Amyloidi on monimutkainen glykoproteiini, joka koostuu kuitu- ja pallomaisia proteiineja läheisesti liittyvät polysakkaridit. Amyloidikertymiä kerääntyä intima ja adventitia verisuonten ja strooman parenkymaalisen elinten, rauhasrakenteet, ja niin edelleen. D. Kun pieniä muutoksia havaitaan amyloidikertymiä vain mikroskooppisella tasolla ja ei johda toiminnallista haittaa. Ilmaisi Amyloidia kertyminen liittyy makroskooppisia muutoksia kyseinen elin (tilavuuden kasvu, rasvainen tai vahamainen view). Lopussa amyloidoosin kehittää tauti ja strooman surkastumista parenkymaaliset elinten ja niiden kliinisesti merkittävää toimintakyvyn vajaatoiminta.
Amyloidoosin luokitus
Mukaisesti syistä erottaa ensisijaisen (idiopaattinen), sekundaarinen (reaktiivinen hankittu), perinnölliset (perhe geneettinen) ja seniili amyloidoosi. Riippuen Primaarivaurio elinten ja järjestelmien eristetty nefropatichesky (munuaisten amyloidoosi) kardiopatichesky (sydämen amyloidoosi), neuropaattinen (amyloidoosi hermosto), gepatopatichesky (amyloidoosi maksa) epinefropatichesky (lisämunuaisen amyloidoosi) ARUD-amyloidoosi ja sekamuotoinen tauti.
Lisäksi kansainvälisen käytännön erottaa paikallisten ja yleistynyt (systeeminen) amyloidoosi. Lokalisoitu muotoja, kehittyy yleensä vanhuksilla kuuluu amyloidoosi Alzheimerin taudissa, tyypin 2 diabetes, hormonaaliset kasvaimet, ihon, virtsarakon ja toiset. Riippuen biokemiallinen koostumus Amyloidisäikeiden keskuudessa systeeminen muotoja amyloidoosin ovat seuraavat tyypit:
- AL — osana fibrillien Ig-kevytketjuja (Waldenstrom-taudin, myelooman, pahanlaatuisten lymfoomien) kanssa;
- AA — muodostuu fibrillien akuutin vaiheen seerumista α-globuliini, samanlainen ominaisuuksiltaan C-reaktiivisen proteiinin (jos kasvain ja reumaattiset taudit jaksollisen ai.);
- Aβ2M — joka koostuu β2-mikroglobuliinin fibrillejä (kroonista munuaisten vajaatoimintaa hemodialyysipotilailla);
- ATTR — osana fibriilejä kuljettaa proteiinit transtyretiiniä (perheen perinnöllisissä ja seniilisissa amyloidoosimuodoissa).
Amyloidoosin oireet
Kliinisiä oireita amyloidoosin eri lajiketta, vakavuuden ja sijainnin amyloidikertymien biokemiallinen koostumus amyloidin, «pituus» sairauden aste elimen toimintahäiriön. Latentti vaihe amyloidoosi kun amyloidikertymiä voidaan havaita vain mikroskoopilla, ei oireita. Kehittämisen ja etenemistä toimintahäiriöitä elimen kasvaa taudin kliinisiä oireita.
Munuaisten amyloidoosilla lievän proteinurian pitkäaikainen vaihe korvataan kehittämällä nefroottista oireyhtymää. Siirtyminen edistyneeseen vaiheeseen voi liittyä transienttiseen intra infektioon, rokotukseen, superjäähdytykseen ja taustalla olevan taudin pahenemiseen. Vähitellen turvotus (ensin jaloissa ja sitten koko kehossa) kehittyy nefrogeenisen valtimon kohonnut verenpaine ja munuaisten vajaatoiminta. Munuaisten laskimotukokset voivat olla tromboosi. Massiivisen proteiinin häviämiseen liittyy hypoproteinemia, hyperfibrinogeneemia, hyperlipidemia, atsotemia. Virtsassa on mikro-, joskus makroematotia, leukosyturia. Yleensä munuaisten amyloidoosiin aikana eristetään varhaiset poistovaihe, edemaattinen vaihe, ureminen (kakaksi) vaihe.
Amyloidoosi sydämen virtaa Tämäntyyppiset rajoittavat sydänlihassairaus tyypillisiä kliinisiä oireita — kardiomegaliaa, rytmihäiriöt, etenevä sydämen vajaatoiminta. Potilaat valittavat hengenahdistus, turvotus, heikkous, esiintyy vain vähän fyysistä rasitusta. Harvinaisempaa amyloidoosi sydämen kehittyy moniherakalvotulehdusta (askites, pleuraeffuusio ja sydänpussinesteen).
Maha-suolikanavan vaurioista amyloidoosi on tunnusomaista amyloidin tunkeutuminen kieli (makroglassiey), ruokatorven (jäykkyys ja motiliteettihäiriöt), vatsa (närästys, pahoinvointi), suolen (ummetus, ripuli, imeytymishäiriö, suolitukos). Saatat kokea ruuansulatuskanavan verenvuotoja eri tasoilla. Kun amyloidi tunkeutumisen maksan kehittyy hepatomegalia, kolestaasi, portaalihypertoniaan. haiman tappio Amyloidoosin yleensä naamioitu krooninen haimatulehdus.
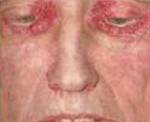
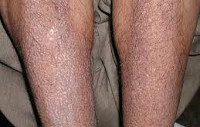
Iho amyloidoosi tapahtuu syntyminen useiden vahamainen plakkien (näppylät, kyhmyt) kasvot, kaula, luonnollinen ihotaipeissa. Näkyy sen kasvot ihomuutokset voivat muistuttaa skleroderma, atooppinen ihottuma tai punajäkälä. Että amyloidin leesioiden tuki- ja liikuntaelinten ovat tyypillisesti kehittäminen symmetrisen polyartriitin, rannekanavaoireyhtymä, jäätynyt olkapää, myopatia. Tietyn tyyppiset amyloidoosin esiintyvien joihin hermostoon voi seurata polyneuropatia, halvaus alaraajojen, päänsärkyä, huimausta, ortostaattinen hypotensio, hikoilu, dementia ja vastaavat. D.
Amyloidoosin diagnoosi
Koska oireenmukaista amyloidoosi voi kohdata eri kliinikot :. Reumatologit, urologit, kardiologeja, gastroenterologeista, neurologit, ihotautilääkärit, lääkärit jne ensisijaisen tärkeää oikean diagnoosin on kattava arviointi kliinisten oireiden ja anamnestisen pitämällä kattava laboratorio- ja instrumentaali tutkimukset.
Erilaisten elinten ja järjestelmien toiminnallisen tilan arvioimiseksi voidaan säätää EchoCG ja EKG; Vatsan ontelon ja munuaisten ultraääni; Ruuansulatus, ruokatorven, vatsa, irrigografia, röntgenkuvaus barium; EGDS, sigmoidoskopia. Amyloidoosi pitäisi ajatella yhdistelmä proteinurian leukocyturia, cylindruria kanssa hypoproteinemia, hyperlipidemia (veren kolesteroli, lipoproteiinit, triglyseridit), hyponatremia ja hypokalsemia, anemiaa, verihiutaleiden määrän laskua. Veren seerumin ja virtsan elektroforeesi mahdollistaa paraproteiinien läsnäolon.
Amyloidoosin lopullinen diagnoosi on mahdollinen amyloidifibrillien havaitsemisen jälkeen vaikuttavissa kudoksissa. Tätä tarkoitusta varten voidaan suorittaa munuais-, kume-, imusolmukkeiden, mahalaukun limakalvon ja peräsuolen biopsia. Amyloidoosin perinnöllisen luonteen luomista helpottaa genealogian varovainen lääketieteellinen geneettinen analyysi.
Amyloidoosin hoito
Täydellisen tiedon puuttuminen taudin etiologiasta ja patogeneesistä aiheuttaa vaikeuksia amyloidoosin hoidossa. Toissijaisessa amyloidoosissa taustataudin aktiivinen hoito on tärkeä. Suositukset ravitsemukselle viittaavat siihen, että taulukon suola ja proteiini on rajoittunut ravinto-ruokavalion sisällyttämiseen. Amyloidoosin oireiden hoito riippuu tiettyjen kliinisten oireiden esiintymisestä ja vakavuudesta.
Patogeenisena hoitona voidaan määrätä 4-aminokinoliinisarjan (klorokiinin), dimetyylisulfoksidin, unitiolin ja kolkisiinin valmisteet. Primaarisen amyloidoosi hoitojakson käyttää sytostaattien ja hormonit (melfolan + prednisoni, vinkristiini, doksorubisiini, deksametasoni). CRF: n kehittymisen myötä hemodialyysi tai peritoneaalidialyysi on osoitettu. Joissakin tapauksissa munuaisen tai maksansiirron kysymys nostetaan.
Entsyymi amyloidoosista
Amyloidoosin kulku on progressiivinen, lähes peruuttamaton luonne. Taudin voi punnita ruokatorven ja mahalaukun amyloidin haavaumat, verenvuoto, maksan vajaatoiminta, diabetes jne. Kroonisen munuaisten vajaatoiminnan kehittymisen myötä potilaiden keskimääräinen elinikä on noin 1 vuosi; sydämen vajaatoiminnan kehittymisen myötä — noin 4 kuukautta. Toissijaisen amyloidoosin ennuste määräytyy taustalla olevan taudin hoidon mahdollisuuden perusteella. Iäkkäillä potilailla havaitaan vaikeampaa amyloidoosia.